Nature ( IF 50.5 ) Pub Date : 2024-06-19 , DOI: 10.1038/s41586-024-07555-1 Marvin Mendel 1 , Teresa M Karl 1 , Jegor Hamm 1 , Sherif J Kaldas 1 , Theresa Sperger 1 , Bhaskar Mondal 1 , Franziska Schoenebeck 1
|
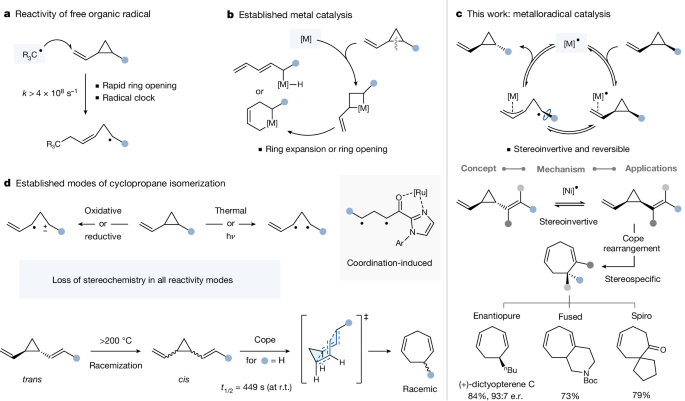
The ever increasing demands for greater sustainability and lower energy usage in chemical processes call for fundamentally new approaches and reactivity principles. In this context, the pronounced prevalence of odd-oxidation states in less precious metals bears untapped potential for fundamentally distinct reactivity modes via metalloradical catalysis1,2,3. Contrary to the well-established reactivity paradigm that organic free radicals, upon addition to a vinylcyclopropane, lead to rapid ring opening under strain release—a transformation that serves widely as a mechanistic probe (radical clock)4 for the intermediacy of radicals5—we herein show that a metal-based radical, that is, a Ni(I) metalloradical, triggers reversible cis/trans isomerization instead of opening. The isomerization proceeds under chiral inversion and, depending on the substitution pattern, occurs at room temperature in less than 5 min, requiring solely the addition of the non-precious catalyst. Our combined computational and experimental mechanistic studies support metalloradical catalysis as origin of this profound reactivity, rationalize the observed stereoinversion and reveal key reactivity features of the process, including its reversibility. These insights enabled the iterative thermodynamic enrichment of enantiopure cis/trans mixtures towards a single diastereomer through multiple Ni(I) catalysis rounds and also extensions to divinylcyclopropanes, which constitute strategic motifs in natural product- and total syntheses6. While the trans-isomer usually requires heating at approximately 200 °C to trigger thermal isomerization under racemization to cis-divinylcyclopropane, which then undergoes facile Cope-type rearrangement, the analogous contra-thermodynamic process is herein shown to proceed under Ni(I) metalloradical catalysis under mild conditions without any loss of stereochemical integrity, enabling a mild and stereochemically pure access to seven-membered rings, fused ring systems and spirocycles.
中文翻译:
乙烯基环丙烷与金属自由基的动态立体突变
对化学过程中更高的可持续性和更低的能源消耗的日益增长的需求需要全新的方法和反应原理。在这种情况下,较少贵金属中奇数氧化态的明显普遍存在,通过金属自由基催化,具有未开发的根本不同的反应模式的潜力1,2,3 。与公认的反应范式相反,有机自由基在添加到乙烯基环丙烷中后,会在应变释放下导致快速开环——这种转变广泛用作自由基中间体的机械探针(自由基时钟) 4 5——我们本文表明金属基自由基,即Ni (I)金属自由基,引发可逆的顺式/反式异构化而不是打开。异构化在手性反转下进行,并且根据取代模式,在室温下发生在不到 5 分钟内,仅需要添加非贵重催化剂。我们结合计算和实验机理研究支持金属自由基催化作为这种深刻反应性的起源,合理化观察到的立体反转并揭示该过程的关键反应特征,包括其可逆性。这些见解使得能够通过多轮 Ni (I)催化对映体纯顺式/反式混合物向单一非对映异构体进行迭代热力学富集,并扩展到二乙烯基环丙烷,这构成了天然产物合成和全合成中的战略主题6 。 虽然反式异构体通常需要在约 200°C 下加热,以在外消旋作用下引发热异构化,生成顺式二乙烯基环丙烷,然后进行简单的 Cope 型重排,但本文显示类似的逆热力学过程在 Ni (I)金属自由基下进行在温和条件下催化,不会损失任何立体化学完整性,能够温和且立体化学纯地获得七元环、稠合环系统和螺环。















































 京公网安备 11010802027423号
京公网安备 11010802027423号