当前位置:
X-MOL 学术
›
Anal. Chim. Acta
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A journey in unraveling the enantiorecognition mechanism of 3,5-dinitrobenzoyl-amino acids with two Cinchona alkaloid-based chiral stationary phases: The power of molecular dynamic simulations
Analytica Chimica Acta ( IF 5.7 ) Pub Date : 2024-05-28 , DOI: 10.1016/j.aca.2024.342791 Ina Varfaj 1 , Magdalena Labikova 2 , Roccaldo Sardella 1 , Hubert Hettegger 3 , Wolfgang Lindner 4 , Michal Kohout 2 , Andrea Carotti 1
Analytica Chimica Acta ( IF 5.7 ) Pub Date : 2024-05-28 , DOI: 10.1016/j.aca.2024.342791 Ina Varfaj 1 , Magdalena Labikova 2 , Roccaldo Sardella 1 , Hubert Hettegger 3 , Wolfgang Lindner 4 , Michal Kohout 2 , Andrea Carotti 1
Affiliation
|
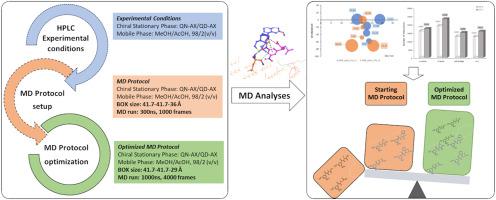
Innovations in computer hardware and software capabilities have paved the way for advances in molecular modelling techniques and methods, leading to an unprecedented expansion of their potential applications. In contrast to the docking technique, which usually identifies the most stable selector-selectand (SO-SA) complex for each enantiomer, the molecular dynamics (MD) technique enables the consideration of a distribution of the SO-SA complexes based on their energy profile. This approach provides a more truthful representation of the processes occurring within the column. However, benchmark procedures and focused guidelines for computational treatment of enantioselectivity at the molecular level are still missing. Twenty-eight molecular dynamics simulations were performed to study the enantiorecognition mechanisms of seven -3,5-dinitrobenzoylated α- and β-amino acids (DNB-AAs), occurring with the two quinine- and quinidine-based (QN-AX and QD-AX) chiral stationary phases (CSPs), under polar-ionic conditions. The MD protocol was optimized in terms of box size, simulation run time, and frame recording frequency. Subsequently, all the trajectories were analyzed by calculating both the type and amount of the interactions engaged by the selectands (SAs) with the two chiral selectors (SOs), as well as the conformational and interaction energy profiles of the formed SA-SO associates. All the MDs were in strict agreement with the experimental enantiomeric elution order and allowed to establish (i) that salt-bridge and H-bond interactions play a pivotal role in the enantiorecognition mechanisms, and (ii) that the π-cation and π-π interactions are the discriminant chemical features between the two SOs in ruling the chiral recognition mechanism. The results of this work clearly demonstrate the high contribution given by MD simulations in the comprehension of the enantiorecognition mechanism with alkaloid-based CSPs. However, from this research endeavor it clearly emerged that the MD protocol optimization is crucial for the quality of the produced results.
中文翻译:

使用两种基于金鸡纳生物碱的手性固定相揭示 3,5-二硝基苯甲酰基氨基酸对映体识别机制的旅程:分子动力学模拟的力量
计算机硬件和软件功能的创新为分子建模技术和方法的进步铺平了道路,从而导致其潜在应用空前扩展。对接技术通常会识别每种对映体最稳定的选择子-选择子 (SO-SA) 复合物,而分子动力学 (MD) 技术则可以根据 SO-SA 复合物的能量分布来考虑其分布。这种方法可以更真实地表示塔内发生的过程。然而,在分子水平上对映选择性计算处理的基准程序和重点指南仍然缺失。进行了 28 次分子动力学模拟,以研究 7 个 -3,5-二硝基苯甲酰化 α- 和 β-氨基酸 (DNB-AA) 与两种奎宁基和奎尼丁基(QN-AX 和 QD)发生的对映体识别机制。 -AX)手性固定相(CSP),在极性离子条件下。 MD 协议在盒子大小、模拟运行时间和帧记录频率方面进行了优化。随后,通过计算选择物 (SA) 与两个手性选择器 (SO) 相互作用的类型和数量,以及形成的 SA-SO 缔合物的构象和相互作用能量分布,对所有轨迹进行了分析。所有 MD 都与实验对映体洗脱顺序严格一致,并允许建立 (i) 盐桥和氢键相互作用在对映体识别机制中发挥关键作用,以及 (ii) π-阳离子和 π- π 相互作用是两个 SO 之间决定手性识别机制的化学特征。 这项工作的结果清楚地证明了 MD 模拟在理解基于生物碱的 CSP 的对映体识别机制方面所做出的巨大贡献。然而,从这项研究工作中可以清楚地看出,MD 方案优化对于生成结果的质量至关重要。
更新日期:2024-05-28
中文翻译:
使用两种基于金鸡纳生物碱的手性固定相揭示 3,5-二硝基苯甲酰基氨基酸对映体识别机制的旅程:分子动力学模拟的力量
计算机硬件和软件功能的创新为分子建模技术和方法的进步铺平了道路,从而导致其潜在应用空前扩展。对接技术通常会识别每种对映体最稳定的选择子-选择子 (SO-SA) 复合物,而分子动力学 (MD) 技术则可以根据 SO-SA 复合物的能量分布来考虑其分布。这种方法可以更真实地表示塔内发生的过程。然而,在分子水平上对映选择性计算处理的基准程序和重点指南仍然缺失。进行了 28 次分子动力学模拟,以研究 7 个 -3,5-二硝基苯甲酰化 α- 和 β-氨基酸 (DNB-AA) 与两种奎宁基和奎尼丁基(QN-AX 和 QD)发生的对映体识别机制。 -AX)手性固定相(CSP),在极性离子条件下。 MD 协议在盒子大小、模拟运行时间和帧记录频率方面进行了优化。随后,通过计算选择物 (SA) 与两个手性选择器 (SO) 相互作用的类型和数量,以及形成的 SA-SO 缔合物的构象和相互作用能量分布,对所有轨迹进行了分析。所有 MD 都与实验对映体洗脱顺序严格一致,并允许建立 (i) 盐桥和氢键相互作用在对映体识别机制中发挥关键作用,以及 (ii) π-阳离子和 π- π 相互作用是两个 SO 之间决定手性识别机制的化学特征。 这项工作的结果清楚地证明了 MD 模拟在理解基于生物碱的 CSP 的对映体识别机制方面所做出的巨大贡献。然而,从这项研究工作中可以清楚地看出,MD 方案优化对于生成结果的质量至关重要。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号