当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Conformational adaptation and large amplitude motions of 1-phenyl-2,2,2-trifluoroethanol with two water molecules: a rotational spectroscopic and ab initio investigation
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-06-12 , DOI: 10.1039/d4cp01516a Colton D Carlson 1 , Jiarui Ma 1 , Mohamad H Al-Jabiri 1 , Aran Insausti 1, 2, 3 , Yunjie Xu 1
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-06-12 , DOI: 10.1039/d4cp01516a Colton D Carlson 1 , Jiarui Ma 1 , Mohamad H Al-Jabiri 1 , Aran Insausti 1, 2, 3 , Yunjie Xu 1
Affiliation
|
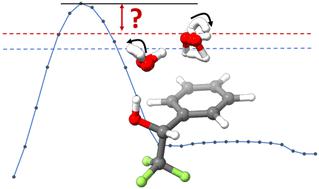
The 1 : 2 adduct of 1-phenyl-2,2,2-trifluoroethanol (PhTFE), a chiral fluoroalcohol, with two water molecules (PhTFE⋯2H2O) was investigated via chirped pulse Fourier-transform microwave (CP-FTMW) spectroscopy and theoretical calculations. A systematic search of the PhTFE⋯2H2O conformational landscape identified 38 stable minima at the B3LYP-D3BJ/def2-TZVPPD level of theory, 27 of which are within an energy window of 10 kJ mol−1 after applying zero-point energy corrections. Rotational spectra of a single PhTFE⋯2H2O conformer along with eight deuterated and three oxygen-18 isotopologues were assigned. Interestingly, the observed PhTFE⋯2H2O conformer contains PhTFE II, the second most stable monomer conformer, and the most stable PhTFE I dihydrate is ca. 4 kJ mol−1 higher in energy. In contrast, PhTFE I⋯H2O was identified experimentally and theoretically as the most stable 1 : 1 conformer. Furthermore, the observed dihydrate structure experiences large amplitude motions connecting three theoretical minima which differ only in which water oxygen lone pairs are involved in the hydrogen-bonds, i.e., the free OH pointing directions. Additionally, the ortho and para-H2O tunnelling splittings were detected and attributed to the interchange water hydrogen atoms which interact with the aromatic part of PhTFE but not for the water interacting with PhTFE hydroxy group. Extensive theoretical modelling was carried out to gain insight into the associated large amplitude motions including tunnelling, supported by the experimental isotopic and tunnelling splitting data.
中文翻译:

1-苯基-2,2,2-三氟乙醇与两个水分子的构象适应和大幅度运动:旋转光谱和从头算研究
通过啁啾脉冲傅里叶变换微波 (CP-FTMW) 研究了 1-苯基-2,2,2-三氟乙醇 (PhTFE)(一种手性氟代醇)与两个水分子 (PhTFE⋯2H 2 O) 的 1:2 加合物光谱学和理论计算。对 PhTFE⋯2H 2 O 构象景观的系统搜索在 B3LYP-D3BJ/def2-TZVPPD 理论水平上确定了 38 个稳定最小值,其中 27 个在应用零点能量校正后位于 10 kJ mol -1的能量窗口内。指定了单个 PhTFE⋯2H 2 O 构象异构体以及八个氘代和三个氧-18 同位素异构体的旋转光谱。有趣的是,观察到的 PhTFE⋯2H 2 O 构象异构体包含第二个最稳定的单体构象异构体 PhTFE II ,而最稳定的 PhTFE I二水合物约为能量高 4 kJ mol -1 。相比之下,PhTFE I ⋯H 2 O 被实验和理论上鉴定为最稳定的 1:1 构象异构体。此外,观察到的二水合物结构经历连接三个理论最小值的大幅运动,其不同之处仅在于氢键中涉及水氧孤对,即自由OH指向方向。 此外,检测到邻位和对位-H 2 O隧道分裂,并将其归因于与PhTFE的芳族部分相互作用的交换水氢原子,而不是与PhTFE羟基相互作用的水氢原子。在实验同位素和隧道分裂数据的支持下,进行了广泛的理论建模,以深入了解相关的大幅运动,包括隧道效应。
更新日期:2024-06-12
中文翻译:
1-苯基-2,2,2-三氟乙醇与两个水分子的构象适应和大幅度运动:旋转光谱和从头算研究
通过啁啾脉冲傅里叶变换微波 (CP-FTMW) 研究了 1-苯基-2,2,2-三氟乙醇 (PhTFE)(一种手性氟代醇)与两个水分子 (PhTFE⋯2H 2 O) 的 1:2 加合物光谱学和理论计算。对 PhTFE⋯2H 2 O 构象景观的系统搜索在 B3LYP-D3BJ/def2-TZVPPD 理论水平上确定了 38 个稳定最小值,其中 27 个在应用零点能量校正后位于 10 kJ mol -1的能量窗口内。指定了单个 PhTFE⋯2H 2 O 构象异构体以及八个氘代和三个氧-18 同位素异构体的旋转光谱。有趣的是,观察到的 PhTFE⋯2H 2 O 构象异构体包含第二个最稳定的单体构象异构体 PhTFE II ,而最稳定的 PhTFE I二水合物约为能量高 4 kJ mol -1 。相比之下,PhTFE I ⋯H 2 O 被实验和理论上鉴定为最稳定的 1:1 构象异构体。此外,观察到的二水合物结构经历连接三个理论最小值的大幅运动,其不同之处仅在于氢键中涉及水氧孤对,即自由OH指向方向。 此外,检测到邻位和对位-H 2 O隧道分裂,并将其归因于与PhTFE的芳族部分相互作用的交换水氢原子,而不是与PhTFE羟基相互作用的水氢原子。在实验同位素和隧道分裂数据的支持下,进行了广泛的理论建模,以深入了解相关的大幅运动,包括隧道效应。





























 京公网安备 11010802027423号
京公网安备 11010802027423号