当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Understanding the Differences of Danusertib’s Residence Time in Aurora Kinases A/B: Dissociation Paths and Key Residues Identified using Conventional and Enhanced Molecular Dynamics Simulations
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-06-10 , DOI: 10.1021/acs.jcim.4c00387 Felipe Bravo-Moraga 1 , Mauricio Bedoya 2, 3 , Ariela Vergara-Jaque 1, 4 , Jans Alzate-Morales 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-06-10 , DOI: 10.1021/acs.jcim.4c00387 Felipe Bravo-Moraga 1 , Mauricio Bedoya 2, 3 , Ariela Vergara-Jaque 1, 4 , Jans Alzate-Morales 1
Affiliation
|
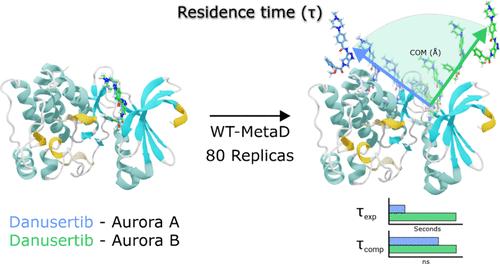
The accurate experimental estimation of protein–ligand systems’ residence time (τ) has become very relevant in drug design projects due to its importance in the last stages of refinement of the drug’s pharmacodynamics and pharmacokinetics. It is now well-known that it is not sufficient to estimate the affinity of a protein-drug complex in the thermodynamic equilibrium process in in vitro experiments (closed systems), where the concentrations of the drug and protein remain constant. On the contrary, it is mandatory to consider the conformational dynamics of the system in terms of the binding and unbinding processes between protein and drugs in in vivo experiments (open systems), where their concentrations are in constant flux. This last model has been proven to dictate much of several drugs’ pharmacological activities in vivo. At the atomistic level, molecular dynamics simulations can explain why some drugs are more effective than others or unveil the molecular aspects that make some drugs work better in one molecular target. Here, the protein kinases Aurora A/B, complexed with its inhibitor Danusertib, were studied using conventional and enhanced molecular dynamics (MD) simulations to estimate the dissociation paths and, therefore, the computational τ values and their comparison with experimental ones. Using classical molecular dynamics (cMD), three differential residues within the Aurora A/B active site, which seems to play an essential role in the observed experimental Danusertib’s residence time against these kinases, were characterized. Then, using WT-MetaD, the relative Danusertib‘s residence times against Aurora A/B kinases were measured in a nanosecond time scale and were compared to those τ values observed experimentally. In addition, the potential dissociation paths of Danusertib in Aurora A and B were characterized, and differences that might be explained by the differential residues in the enzyme’s active sites were found. In perspective, it is expected that this computational protocol can be applied to other protein–ligand complexes to understand, at the molecular level, the differences in residence times and amino acids that may contribute to it.
中文翻译:

了解 Danusertib 在 Aurora 激酶 A/B 中的停留时间差异:使用常规和增强型分子动力学模拟鉴定的解离路径和关键残基
蛋白质-配体系统停留时间 (τ) 的准确实验估计在药物设计项目中变得非常重要,因为它在药物药效学和药代动力学改进的最后阶段非常重要。现在众所周知,在体外实验(封闭系统)中,在药物和蛋白质的浓度保持恒定的情况下,在热力学平衡过程中估计蛋白质-药物复合物的亲和力是不够的。相反,在体内实验(开放系统)中,必须根据蛋白质和药物之间的结合和解结合过程来考虑系统的构象动力学,其中它们的浓度处于恒定变化中。最后一种模型已被证明决定了几种药物在体内的大部分药理活性。在原子水平上,分子动力学模拟可以解释为什么某些药物比其他药物更有效,或者揭示使某些药物在一个分子靶标中效果更好的分子方面。在这里,使用常规和增强分子动力学 (MD) 模拟研究了与其抑制剂 Danusertib 复合的蛋白激酶 Aurora A/B,以估计解离路径,从而估计计算 τ 值及其与实验值的比较。使用经典分子动力学 (cMD),表征了 Aurora A/B 活性位点内的三个差异残基,这似乎在观察到的实验 Danusertib 对这些激酶的停留时间中起着重要作用。然后,使用 WT-MetaD,以纳秒时间尺度测量 Danusertib 对 Aurora A/B 激酶的相对停留时间,并与实验观察到的 τ 值进行比较。 此外,还表征了 Aurora A 和 B 中 Danusertib 的潜在解离途径,并发现了可能由酶活性位点中的差异残基解释的差异。从长远来看,预计该计算方案可以应用于其他蛋白质-配体复合物,以在分子水平上了解可能导致它的停留时间和氨基酸的差异。
更新日期:2024-06-10
中文翻译:
了解 Danusertib 在 Aurora 激酶 A/B 中的停留时间差异:使用常规和增强型分子动力学模拟鉴定的解离路径和关键残基
蛋白质-配体系统停留时间 (τ) 的准确实验估计在药物设计项目中变得非常重要,因为它在药物药效学和药代动力学改进的最后阶段非常重要。现在众所周知,在体外实验(封闭系统)中,在药物和蛋白质的浓度保持恒定的情况下,在热力学平衡过程中估计蛋白质-药物复合物的亲和力是不够的。相反,在体内实验(开放系统)中,必须根据蛋白质和药物之间的结合和解结合过程来考虑系统的构象动力学,其中它们的浓度处于恒定变化中。最后一种模型已被证明决定了几种药物在体内的大部分药理活性。在原子水平上,分子动力学模拟可以解释为什么某些药物比其他药物更有效,或者揭示使某些药物在一个分子靶标中效果更好的分子方面。在这里,使用常规和增强分子动力学 (MD) 模拟研究了与其抑制剂 Danusertib 复合的蛋白激酶 Aurora A/B,以估计解离路径,从而估计计算 τ 值及其与实验值的比较。使用经典分子动力学 (cMD),表征了 Aurora A/B 活性位点内的三个差异残基,这似乎在观察到的实验 Danusertib 对这些激酶的停留时间中起着重要作用。然后,使用 WT-MetaD,以纳秒时间尺度测量 Danusertib 对 Aurora A/B 激酶的相对停留时间,并与实验观察到的 τ 值进行比较。 此外,还表征了 Aurora A 和 B 中 Danusertib 的潜在解离途径,并发现了可能由酶活性位点中的差异残基解释的差异。从长远来看,预计该计算方案可以应用于其他蛋白质-配体复合物,以在分子水平上了解可能导致它的停留时间和氨基酸的差异。





























 京公网安备 11010802027423号
京公网安备 11010802027423号