npj Computational Materials ( IF 9.4 ) Pub Date : 2024-05-30 , DOI: 10.1038/s41524-024-01256-z Gaurav Deshmukh , Noah J. Wichrowski , Nikolaos Evangelou , Pushkar G. Ghanekar , Siddharth Deshpande , Ioannis G. Kevrekidis , Jeffrey Greeley
|
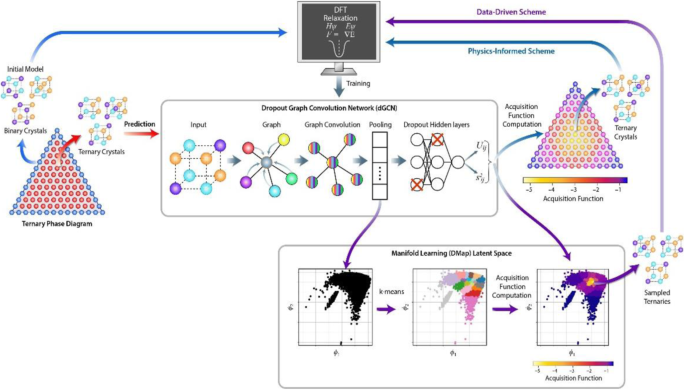
Machine learning models with uncertainty quantification have recently emerged as attractive tools to accelerate the navigation of catalyst design spaces in a data-efficient manner. Here, we combine active learning with a dropout graph convolutional network (dGCN) as a surrogate model to explore the complex materials space of high-entropy alloys (HEAs). We train the dGCN on the formation energies of disordered binary alloy structures in the Pd-Pt-Sn ternary alloy system and improve predictions on ternary structures by performing reduced optimization of the formation free energy, the target property that determines HEA stability, over ensembles of ternary structures constructed based on two coordinate systems: (a) a physics-informed ternary composition space, and (b) data-driven coordinates discovered by the Diffusion Maps manifold learning scheme. Both reduced optimization techniques improve predictions of the formation free energy in the ternary alloy space with a significantly reduced number of DFT calculations compared to a high-fidelity model. The physics-based scheme converges to the target property in a manner akin to a depth-first strategy, whereas the data-driven scheme appears more akin to a breadth-first approach. Both sampling schemes, coupled with our acquisition function, successfully exploit a database of DFT-calculated binary alloy structures and energies, augmented with a relatively small number of ternary alloy calculations, to identify stable ternary HEA compositions and structures. This generalized framework can be extended to incorporate more complex bulk and surface structural motifs, and the results demonstrate that significant dimensionality reduction is possible in thermodynamic sampling problems when suitable active learning schemes are employed.
中文翻译:
主动学习三元合金结构和能量
具有不确定性量化的机器学习模型最近已成为有吸引力的工具,可以以数据有效的方式加速催化剂设计空间的导航。在这里,我们将主动学习与 dropout 图卷积网络(dGCN)相结合作为替代模型来探索高熵合金(HEA)的复杂材料空间。我们在 Pd-Pt-Sn 三元合金系统中无序二元合金结构的形成能上训练 dGCN,并通过对形成自由能(决定 HEA 稳定性的目标属性)进行减少优化来改进对三元结构的预测。基于两个坐标系构建的三元结构:(a)物理信息三元组成空间,以及(b)由扩散图流形学习方案发现的数据驱动坐标。与高保真模型相比,这两种简化的优化技术都显着减少了 DFT 计算数量,从而改进了三元合金空间中形成自由能的预测。基于物理的方案以类似于深度优先策略的方式收敛到目标属性,而数据驱动的方案似乎更类似于广度优先方法。两种采样方案与我们的采集功能相结合,成功地利用了 DFT 计算的二元合金结构和能量数据库,并通过相对少量的三元合金计算进行了扩充,以识别稳定的三元 HEA 成分和结构。这个广义框架可以扩展以包含更复杂的体积和表面结构图案,结果表明,当采用合适的主动学习方案时,热力学采样问题的维数可以显着降低。










































 京公网安备 11010802027423号
京公网安备 11010802027423号