当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A systematic DFT study of structure and electronic properties of titanium dioxide
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-05-24 , DOI: 10.1002/jcc.27376 Asma Marzouk 1 , Konstantinos D. Papavasileiou 2 , Loukas D. Peristeras 2 , Leendert Bezemer 3 , Alexander P. van Bavel 4 , Prathamesh M. Shenai 5 , Ioannis G. Economou 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-05-24 , DOI: 10.1002/jcc.27376 Asma Marzouk 1 , Konstantinos D. Papavasileiou 2 , Loukas D. Peristeras 2 , Leendert Bezemer 3 , Alexander P. van Bavel 4 , Prathamesh M. Shenai 5 , Ioannis G. Economou 1
Affiliation
|
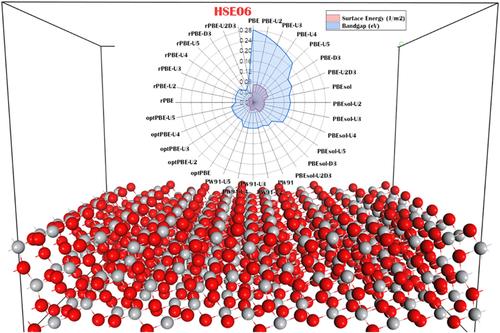
DFT functionals are of paramount importance for an accurate electronic and structural description of transition metal systems. In this work, a systematic analysis using some well‐known and commonly used DFT functionals is performed. A comparison of the structural and energetic parameters calculated with the available experimental data is made in order to find the adequate functional for an accurate description of the TiO2 bulk and surface of both anatase and rutile structures. In the absence of experimental data on the surface energy, the theoretical predictions obtained using the high‐accuracy HSE06 functional were used as a reference to compare against the surface energy values calculated with the other DFT functionals. A clear improvement in the electronic description of both anatase and rutile was observed by introducing the Hubbard U correction term to PBE, PW91, and OptPBE functionals. The OptPBE‐U 4 functional was found to offer a good compromise between accurately describing the structural and electronic properties of titania.
中文翻译:

二氧化钛结构和电子性质的系统DFT研究
DFT 泛函对于过渡金属系统的精确电子和结构描述至关重要。在这项工作中,使用一些众所周知且常用的 DFT 泛函进行了系统分析。对与现有实验数据计算出的结构和能量参数进行了比较,以便找到准确描述锐钛矿和金红石结构的 TiO2 体积和表面的足够函数。在缺乏表面能实验数据的情况下,使用高精度 HSE06 泛函获得的理论预测被用作参考,与其他 DFT 泛函计算的表面能值进行比较。通过将 Hubbard U 校正项引入 PBE、PW91 和 OptPBE 泛函,观察到锐钛矿和金红石的电子描述有明显改进。 OptPBE-U4 泛函被发现可以在准确描述二氧化钛的结构和电子特性之间提供良好的折衷。
更新日期:2024-05-24
中文翻译:
二氧化钛结构和电子性质的系统DFT研究
DFT 泛函对于过渡金属系统的精确电子和结构描述至关重要。在这项工作中,使用一些众所周知且常用的 DFT 泛函进行了系统分析。对与现有实验数据计算出的结构和能量参数进行了比较,以便找到准确描述锐钛矿和金红石结构的 TiO2 体积和表面的足够函数。在缺乏表面能实验数据的情况下,使用高精度 HSE06 泛函获得的理论预测被用作参考,与其他 DFT 泛函计算的表面能值进行比较。通过将 Hubbard U 校正项引入 PBE、PW91 和 OptPBE 泛函,观察到锐钛矿和金红石的电子描述有明显改进。 OptPBE-U4 泛函被发现可以在准确描述二氧化钛的结构和电子特性之间提供良好的折衷。











































 京公网安备 11010802027423号
京公网安备 11010802027423号