Communications Biology ( IF 5.2 ) Pub Date : 2024-05-13 , DOI: 10.1038/s42003-024-06238-x
Thomas G Hayhow 1 , Beth Williamson 1 , Mandy Lawson 1 , Natalie Cureton 1 , Erin L Braybrooke 1 , Andrew Campbell 2 , Rodrigo J Carbajo 1 , Azadeh Cheraghchi-Bashi 1 , Elisabetta Chiarparin 1 , Coura R Diène 1 , Charlene Fallan 1 , David I Fisher 3 , Frederick W Goldberg 1 , Lorna Hopcroft 1 , Philip Hopcroft 3 , Anne Jackson 3 , Jason G Kettle 1 , Teresa Klinowska 1 , Ulrike Künzel 3 , Gillian Lamont 1 , Hilary J Lewis 1 , Gareth Maglennon 1 , Scott Martin 1 , Pablo Morentin Gutierrez 1 , Christopher J Morrow 1 , Myria Nikolaou 1 , J Willem M Nissink 1 , Patrick O'Shea 3 , Radoslaw Polanski 3 , Markus Schade 1 , James S Scott 1 , Aaron Smith 1 , Judith Weber 1 , Joanne Wilson 1 , Bin Yang 4 , Claire Crafter 1
|
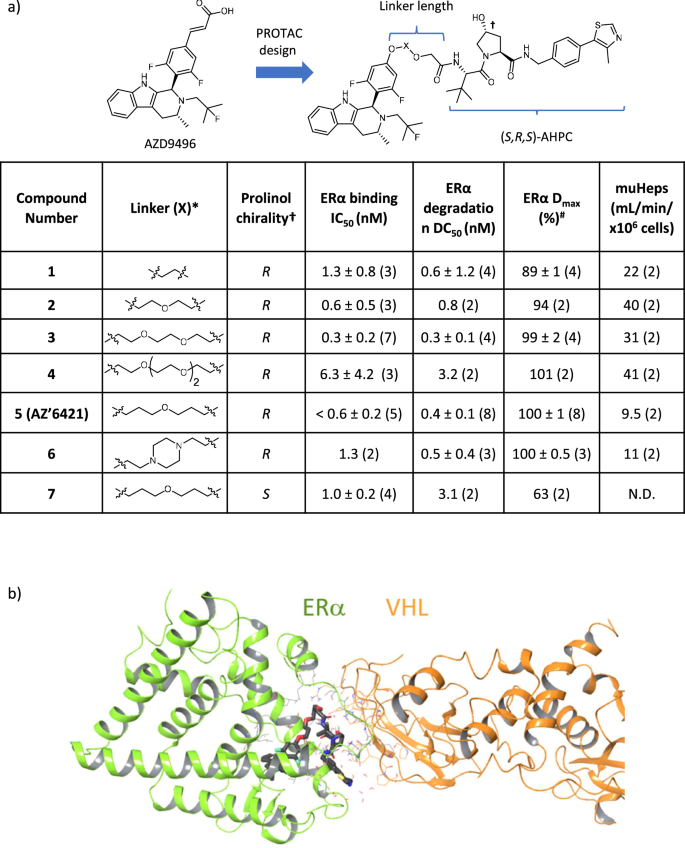
Targeting the estrogen receptor alpha (ERα) pathway is validated in the clinic as an effective means to treat ER+ breast cancers. Here we present the development of a VHL-targeting and orally bioavailable proteolysis-targeting chimera (PROTAC) degrader of ERα. In vitro studies with this PROTAC demonstrate excellent ERα degradation and ER antagonism in ER+ breast cancer cell lines. However, upon dosing the compound in vivo we observe an in vitro-in vivo disconnect. ERα degradation is lower in vivo than expected based on the in vitro data. Investigation into potential causes for the reduced maximal degradation reveals that metabolic instability of the PROTAC linker generates metabolites that compete for binding to ERα with the full PROTAC, limiting degradation. This observation highlights the requirement for metabolically stable PROTACs to ensure maximal efficacy and thus optimisation of the linker should be a key consideration when designing PROTACs.
中文翻译:
口服 ERɑ VHL-PROTAC 的代谢驱动的体外/体内断开
靶向雌激素受体 α (ERα) 通路已在临床上得到验证,是治疗 ER+ 乳腺癌的有效手段。在这里,我们介绍了 ERα 的 VHL 靶向和口服生物可利用的蛋白水解靶向嵌合体 (PROTAC) 降解剂的开发。使用该 PROTAC 的体外研究表明,在 ER+ 乳腺癌细胞系中具有优异的 ERα 降解和 ER 拮抗作用。然而,在体内给药化合物时,我们观察到体外-体内脱节。ERα 在体内的降解低于基于体外数据的预期。对最大降解减少潜在原因的调查表明,PROTAC 接头的代谢不稳定性会产生代谢物,这些代谢物与完整的 PROTAC 竞争与 ERα 结合,从而限制降解。这一观察结果强调了代谢稳定的 PROTAC 以确保最大疗效的要求,因此在设计 PROTAC 时,优化接头应是一个关键考虑因素。






























 京公网安备 11010802027423号
京公网安备 11010802027423号