当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
SCC-DFTB 与标准和混合 DFT 的基准研究,用于模拟热电应用二维 MOF 的电子特性
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-05-06 , DOI: 10.1021/acs.jctc.3c01405 Masoumeh Mahmoudi Gahrouei 1 , Nikiphoros Vlastos 1 , Ransell D'Souza 2 , Emmanuel C Odogwu 1 , Laura de Sousa Oliveira 1
Affiliation
|
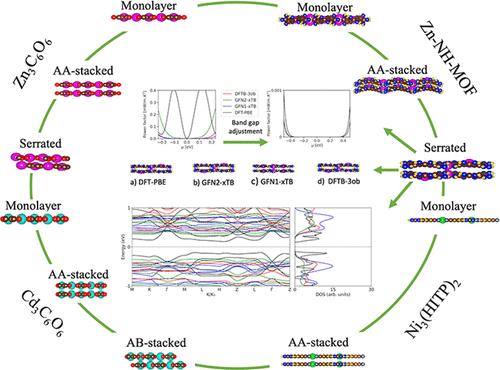
最近的研究表明,金属有机框架(MOF)具有作为热电材料的潜力,并且该主题受到越来越多的关注。该项目的主要动机是加深我们对 MOF 热电特性的了解,并找到哪种可用的自洽电荷密度功能紧密结合 (SCC-DFTB) 方法可以最好地预测 MOF 的电子特性(至少是趋势)比标准密度泛函理论 (DFT) 的计算成本更低。在这项工作中,单层、锯齿状、AA 堆叠和/或 AB 堆叠 Zn 3 C 6 O 6 、Cd 3 C 6 O 6 、Zn-NH-MOF 的电子特性 — 之前没有计算过热电性能存在 — 并且 Ni 3 (HITP) 2 MOF 使用 DFT-PBE、DFT-HSE06、GFN1-xTB、GFN2-xTB 和 DFTB-3ob/mio 进行建模。不同方法和几何结构对能带结构、态密度及其相对轨道贡献以及电导率、塞贝克系数和功率因数进行了比较。我们的结果表明 GFN-xTB 足以预测 MOF 的能带结构形状和态密度,但不能预测带隙。我们的计算进一步表明,Zn 3 C 6 O 6 、Cd 3 C 6 O 6和 Zn-NH-MOF 具有比性能最高的合成 MOF 之一 Ni 3 (HITP) 2更高的功率因数值,因此很有前景用于热电应用。
"点击查看英文标题和摘要"






























 京公网安备 11010802027423号
京公网安备 11010802027423号