当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
GraphEGFR: Multi-task and transfer learning based on molecular graph attention mechanism and fingerprints improving inhibitor bioactivity prediction for EGFR family proteins on data scarcity
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-05-07 , DOI: 10.1002/jcc.27388 Bundit Boonyarit 1 , Nattawin Yamprasert 2 , Pawit Kaewnuratchadasorn 3 , Jiramet Kinchagawat 1 , Chanatkran Prommin 1 , Thanyada Rungrotmongkol 4, 5 , Sarana Nutanong 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-05-07 , DOI: 10.1002/jcc.27388 Bundit Boonyarit 1 , Nattawin Yamprasert 2 , Pawit Kaewnuratchadasorn 3 , Jiramet Kinchagawat 1 , Chanatkran Prommin 1 , Thanyada Rungrotmongkol 4, 5 , Sarana Nutanong 1
Affiliation
|
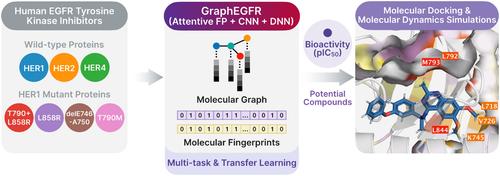
The proteins within the human epidermal growth factor receptor (EGFR) family, members of the tyrosine kinase receptor family, play a pivotal role in the molecular mechanisms driving the development of various tumors. Tyrosine kinase inhibitors, key compounds in targeted therapy, encounter challenges in cancer treatment due to emerging drug resistance mutations. Consequently, machine learning has undergone significant evolution to address the challenges of cancer drug discovery related to EGFR family proteins. However, the application of deep learning in this area is hindered by inherent difficulties associated with small-scale data, particularly the risk of overfitting. Moreover, the design of a model architecture that facilitates learning through multi-task and transfer learning, coupled with appropriate molecular representation, poses substantial challenges. In this study, we introduce GraphEGFR, a deep learning regression model designed to enhance molecular representation and model architecture for predicting the bioactivity of inhibitors against both wild-type and mutant EGFR family proteins. GraphEGFR integrates a graph attention mechanism for molecular graphs with deep and convolutional neural networks for molecular fingerprints. We observed that GraphEGFR models employing multi-task and transfer learning strategies generally achieve predictive performance comparable to existing competitive methods. The integration of molecular graphs and fingerprints adeptly captures relationships between atoms and enables both global and local pattern recognition. We further validated potential multi-targeted inhibitors for wild-type and mutant HER1 kinases, exploring key amino acid residues through molecular dynamics simulations to understand molecular interactions. This predictive model offers a robust strategy that could significantly contribute to overcoming the challenges of developing deep learning models for drug discovery with limited data and exploring new frontiers in multi-targeted kinase drug discovery for EGFR family proteins.
中文翻译:

GraphEGFR:基于分子图注意机制和指纹的多任务和迁移学习,改善数据稀缺情况下 EGFR 家族蛋白的抑制剂生物活性预测
人表皮生长因子受体 (EGFR) 家族中的蛋白质是酪氨酸激酶受体家族的成员,在驱动各种肿瘤发展的分子机制中发挥着关键作用。酪氨酸激酶抑制剂是靶向治疗的关键化合物,由于新出现的耐药突变,在癌症治疗中遇到了挑战。因此,机器学习经历了重大发展,以应对与 EGFR 家族蛋白相关的癌症药物发现的挑战。然而,深度学习在这一领域的应用受到小规模数据固有困难的阻碍,特别是过度拟合的风险。此外,通过多任务和迁移学习促进学习的模型架构的设计,加上适当的分子表示,提出了巨大的挑战。在这项研究中,我们引入了 GraphEGFR,这是一种深度学习回归模型,旨在增强分子表征和模型架构,以预测针对野生型和突变型 EGFR 家族蛋白的抑制剂的生物活性。 GraphEGFR 将分子图的图注意力机制与分子指纹的深度卷积神经网络集成在一起。我们观察到,采用多任务和迁移学习策略的 GraphEGFR 模型通常可以实现与现有竞争方法相当的预测性能。分子图和指纹的集成巧妙地捕获了原子之间的关系,并实现了全局和局部模式识别。我们进一步验证了野生型和突变型 HER1 激酶的潜在多靶点抑制剂,通过分子动力学模拟探索关键氨基酸残基以了解分子相互作用。 这种预测模型提供了一种强大的策略,可以极大地帮助克服利用有限数据开发药物发现深度学习模型的挑战,并探索 EGFR 家族蛋白多靶点激酶药物发现的新领域。
更新日期:2024-05-07
中文翻译:
GraphEGFR:基于分子图注意机制和指纹的多任务和迁移学习,改善数据稀缺情况下 EGFR 家族蛋白的抑制剂生物活性预测
人表皮生长因子受体 (EGFR) 家族中的蛋白质是酪氨酸激酶受体家族的成员,在驱动各种肿瘤发展的分子机制中发挥着关键作用。酪氨酸激酶抑制剂是靶向治疗的关键化合物,由于新出现的耐药突变,在癌症治疗中遇到了挑战。因此,机器学习经历了重大发展,以应对与 EGFR 家族蛋白相关的癌症药物发现的挑战。然而,深度学习在这一领域的应用受到小规模数据固有困难的阻碍,特别是过度拟合的风险。此外,通过多任务和迁移学习促进学习的模型架构的设计,加上适当的分子表示,提出了巨大的挑战。在这项研究中,我们引入了 GraphEGFR,这是一种深度学习回归模型,旨在增强分子表征和模型架构,以预测针对野生型和突变型 EGFR 家族蛋白的抑制剂的生物活性。 GraphEGFR 将分子图的图注意力机制与分子指纹的深度卷积神经网络集成在一起。我们观察到,采用多任务和迁移学习策略的 GraphEGFR 模型通常可以实现与现有竞争方法相当的预测性能。分子图和指纹的集成巧妙地捕获了原子之间的关系,并实现了全局和局部模式识别。我们进一步验证了野生型和突变型 HER1 激酶的潜在多靶点抑制剂,通过分子动力学模拟探索关键氨基酸残基以了解分子相互作用。 这种预测模型提供了一种强大的策略,可以极大地帮助克服利用有限数据开发药物发现深度学习模型的挑战,并探索 EGFR 家族蛋白多靶点激酶药物发现的新领域。










































 京公网安备 11010802027423号
京公网安备 11010802027423号