Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2024-04-24 , DOI: 10.1002/adsc.202400316 Jeanne Fichez 1 , Maria Ivana Lapuh 1 , Lina Truong 1 , Hassan Oulyadi 2 , Floris Buttard 1 , Tatiana Besset 3
|
Introduction
The functionalization of inert C−H bonds by transition metal catalysis has become a tool of paramount importance in organic chemistry for the creation of new carbon−carbon and carbon−heteroatom bonds.1 Naturally, this strategy was used for the synthesis of complex molecular scaffolds, natural products and was applied for late-stage functionalizations of various compounds including bioactive ingredients.2 To cope with the difficulty of selectively functionalizing a targeted C−H bond, one common approach relies on the use of directing groups. These later can coordinate the metal catalyst and place it close to the targeted C−H bond via the formation of a suitable metallacycle.1, 3 More recently, molecular template- or secondary interactions-directed approaches have also been developed to perform the activation of C−H bonds located at distal positions.4 A complementary strategy relies on non-directed C−H activation approaches, circumventing the drawbacks related to the installation and the cleavage of the directing group, but sometimes at the expense of regioselectivity.5
Among these various C−H functionalization approaches, Cross Dehydrogenative Coupling (CDC) reactions are of high interest from non-functionalized substrates6, 7, 8 with the venerable Fujiwara-Moritani olefination as a flagship reaction.9 In 2009, the group of Yu developed the meta-selective C−H olefination of highly electron-deficient arenes using a bulky 2,6-dialkylpyridine ligand L1 (Scheme 1).10a The role of this monodentate nitrogen-based ligand was critical to enhance the catalytic activity of palladium complexes and to tune the regioselectivity as illustrated in other palladium-catalyzed non-directed C−H bond arylation, acetoxylation and olefination reactions.10 Since these pioneering works, several ligands have been designed to improve and modulate the reactivity and selectivity of the Fujiwara-Moritani reaction. Among them, the 3,5-dichloropyridine L2,11 bidentate 2-hydroxyphenanthroline L3a12a and [2,2’-bipyridin]-6(1H)-one L3b,12b pyrazole-substituted 2-pyridone L4,13 or the weakly coordinating bidentate thioethercarboxylic acid ligand L514 are particularly efficient. Thanks to these major advances, arene-limited transformations were then possible using for instance the 2-pyridone L6[15] or a combination of the pyridine L7 and N-acetylglycine.16

Ligand-accelerated non-directed Fujiwara-Moritani olefinations. L1: Yu et al., 2009;10a L2: Sanford et al., 2012;11 L3a: Duan et al., 2014;12a L3b: Albéniz and Villalba, 2021;12b L4: Joo et al., 2023;13 L5: Fernández-Ibáñez et al., 2017;14 L6: Yu et al., 2017;15 L6: Van Gemmeren et al., 2018.16
In an effort to valorize raw materials for more sustainable chemical transformations, chlorobenzene, a feedstock chemical, was olefinated (used either in a stoichiometric quantity or as a solvent) in several above-mentioned reports,12, 13, 15 as well as in early works employing catalytic systems without pyridine ligands.[17−19] Recently, copper-containing heterometallic complexes of rhodium and palladium have also been reported for the olefination of a variety of arenes, including chlorobenzene.20 The regioselectivity observed in these works always favored an olefination at the meta over the ortho-to-chlorine position (Scheme 2).

Non-directed Fujiwara-Moritani olefinations on chlorobenzene: State of the Art and Present work.
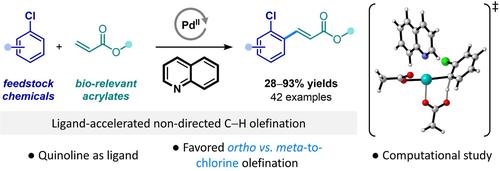
However, when polysubstituted chloroarenes were used, an unusual ortho-to-chlorine selectivity was already observed, although restricted to two examples.11, 16 Maiti and co-workers also reported a photo-induced para-selective olefination of chlorobenzene by merging palladium(II) and organo-photocatalysis.21 Intrigued by these singular results and since the control and modulation of the site-selectivity in transition metal non-directed C−H olefinations are still identified as major challenges to be addressed, the quest for innovative solutions is of utmost importance. As a result, we wondered whether tuning the nature of ligands could be used to switch the regioselectivity. Herein, we report a Fujiwara-Moritani olefination on chlorobenzene derivatives favoring the functionalization at the ortho over meta-to-chlorine position by using the simple and commercially available quinoline as a ligand.
中文翻译:
非定向钯催化氯苯衍生物 C−H 烯化反应中的选择性开关
介绍
通过过渡金属催化对惰性 C−H 键进行官能化已成为有机化学中用于创建新的碳−碳和碳−杂原子键的最重要的工具。 1 自然地,该策略用于复杂分子支架、天然产物的合成,并应用于包括生物活性成分在内的各种化合物的后期功能化。 2 为了解决选择性功能化目标 C−H 键的困难,一种常见的方法依赖于使用导向基团。这些随后可以配位金属催化剂,并通过形成合适的金属环将其放置在目标 CH 键附近。 1, 3 最近,还开发了分子模板或二次相互作用导向的方法来激活位于远端位置的 CH 键。 4 补充策略依赖于非定向 C−H 激活方法,避免了与定向基团安装和裂解相关的缺点,但有时会牺牲区域选择性。 5
在这些不同的CH官能化方法中,交叉脱氢偶联(CDC)反应受到非官能化底物 6, 7, 8 的高度关注,其中古老的Fujiwara-Moritani烯化反应作为旗舰反应。 9 2009年,Yu课题组利用大体积的2,6-二烷基吡啶配体L1开发了高度缺电子芳烃的间位选择性CH烯化反应(方案1)。 10a 这种单齿氮基配体的作用对于增强钯配合物的催化活性和调整区域选择性至关重要,如其他钯催化的非定向 C−H 键芳基化、乙酰氧基化和烯化反应所示。反应。 10 自从这些开创性工作以来,已经设计了几种配体来改善和调节藤原-森谷反应的反应性和选择性。其中,3,5-二氯吡啶L2, 11 二齿2-羟基菲咯啉L3a 12a 和[2,2'-联吡啶]-6(1H)-酮L3b, 12b 吡唑取代的2-吡啶酮L4、 13 或弱配位二齿硫醚羧酸配体L5 14 特别有效。由于这些重大进展,使用 2-吡啶酮 L6 [ 15] 或吡啶 L7 和 N-乙酰甘氨酸的组合可以实现芳烃限制转化。 16
在图查看器中打开PowerPoint
配体加速非定向 Fujiwara-Moritani 烯化。 L1:Yu 等,2009; 10a L2:Sanford 等人,2012; 11 L3a:段等人,2014; 12a L3b:阿尔贝尼兹和维拉尔巴,2021 年; 12b L4:Joo 等人,2023; 13 L5:Fernández-Ibáñez 等人,2017 年; 14 L6:Yu 等,2017; 15 L6:Van Gemmeren 等人,2018。 16
为了稳定原材料以实现更可持续的化学转化,在上述几份报告中,氯苯(一种原料化学品)被烯化(以化学计量使用或作为溶剂), 12, 13, 15 以及在早期的工作中使用没有吡啶配体的催化系统。 [17−19] 最近,铑和钯的含铜异金属配合物也被报道用于多种芳烃的烯化,包括氯苯。 20 在这些工作中观察到的区域选择性总是有利于间位烯化而不是邻位到氯位(方案 2)。
在图查看器中打开PowerPoint
氯苯的非定向 Fujiwara-Moritani 烯化:最新技术和当前工作。
然而,当使用多取代的氯芳烃时,已经观察到不寻常的邻氯选择性,尽管仅限于两个例子。 11, 16 Maiti 和同事还报道了通过结合钯 (II) 和有机光催化来光诱导氯苯的对位选择性烯化。 21 对这些奇异的结果很感兴趣,并且由于过渡金属非定向 C−H 烯化反应中位点选择性的控制和调节仍然被认为是需要解决的主要挑战,因此寻求创新解决方案是迫切需要解决的问题。最重要的。因此,我们想知道是否可以通过调整配体的性质来改变区域选择性。在此,我们报道了氯苯衍生物的 Fujiwara-Moritani 烯化反应,通过使用简单且市售的喹啉作为配体,有利于邻位而不是间位氯位置的官能化。









































 京公网安备 11010802027423号
京公网安备 11010802027423号