当前位置:
X-MOL 学术
›
Bioorgan. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The new N2, N4-diphenylpyridine-2,4-diamine deuterated derivatives as EGFR inhibitors to overcome C797S-mediated resistance
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2024-03-26 , DOI: 10.1016/j.bioorg.2024.107313 Jiadai Liu 1 , Wenyan Nie 2 , Haoran Nie 2 , Han Yao 3 , Yuanyuan Ren 3 , Longcai Cao 1 , Jiaqi Qiu 2 , Mengxuan Wang 2 , Xingshu Li 3 , Baijiao An 2 , Xian Jia 1
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2024-03-26 , DOI: 10.1016/j.bioorg.2024.107313 Jiadai Liu 1 , Wenyan Nie 2 , Haoran Nie 2 , Han Yao 3 , Yuanyuan Ren 3 , Longcai Cao 1 , Jiaqi Qiu 2 , Mengxuan Wang 2 , Xingshu Li 3 , Baijiao An 2 , Xian Jia 1
Affiliation
|
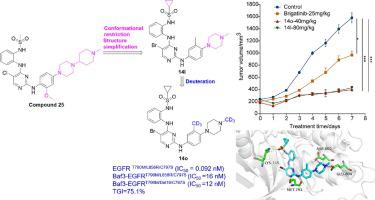
A series of new deuterated and non-deuterated , -diphenylpyridine − 2,4-diamine derivatives were synthesized and evaluated as EGFR C797S-mediated resistance inhibitors. Most of these compounds exhibited potent antiproliferative activity against Baf3-EGFR and Baf3-EGFR cancel cell lines, with IC values in the nanomolar concentration range. Among them, compound represented the most active compound with IC values of 8–11 nM. Interestingly, metabolic stability assay with rat liver microsomes indicated that the half-life of the deuterated derivative was significantly increased compared to that of . In xenograft mice models, 1 inhibited tumor growth with excellent inhibitory rate of 75.1 % at the dosage of 40 mg/kg, comparing 73.2 % of the TGI with its non-deuterated compound , at a dosage of 80 mg/kg. Mechanism studies revealed that was a potent EGFR and EGFR kinase inhibitor, which could downregulate the protein phosphorylation of EGFR and m-TOR signaling pathways, arrest cell cycle at G2/M phase by affecting the expression of CDC25C, and promote cell apoptosis by regulating the expression of cleaved caspase-3. In summary, could serve as a promising deuterated compound for the development of highly efficient anticancer agents.
中文翻译:

新型 N2,N4-二苯基吡啶-2,4-二胺氘代衍生物作为 EGFR 抑制剂,克服 C797S 介导的耐药性
合成了一系列新的氘代和非氘代 , -二苯基吡啶 - 2,4-二胺衍生物,并作为 EGFR C797S 介导的耐药抑制剂进行了评估。大多数这些化合物对 Baf3-EGFR 和 Baf3-EGFR 取消细胞系表现出有效的抗增殖活性,IC 值在纳摩尔浓度范围内。其中,化合物是最活跃的化合物,IC 值为 8-11 nM。有趣的是,大鼠肝微粒体的代谢稳定性测定表明,与 相比,氘代衍生物的半衰期显着增加。在异种移植小鼠模型中,1 在 40 mg/kg 剂量下抑制肿瘤生长,抑制率为 75.1%,而 TGI 与其非氘化化合物在 80 mg/kg 剂量下的抑制率为 73.2%。机制研究表明,它是一种有效的 EGFR 和 EGFR 激酶抑制剂,可下调 EGFR 和 m-TOR 信号通路的蛋白磷酸化,通过影响 CDC25C 的表达将细胞周期阻滞在 G2/M 期,并通过调节 CDC25C 的表达来促进细胞凋亡。切割的 caspase-3 的表达。总之,可以作为一种有前途的氘化化合物,用于开发高效抗癌剂。
更新日期:2024-03-26
中文翻译:
新型 N2,N4-二苯基吡啶-2,4-二胺氘代衍生物作为 EGFR 抑制剂,克服 C797S 介导的耐药性
合成了一系列新的氘代和非氘代 , -二苯基吡啶 - 2,4-二胺衍生物,并作为 EGFR C797S 介导的耐药抑制剂进行了评估。大多数这些化合物对 Baf3-EGFR 和 Baf3-EGFR 取消细胞系表现出有效的抗增殖活性,IC 值在纳摩尔浓度范围内。其中,化合物是最活跃的化合物,IC 值为 8-11 nM。有趣的是,大鼠肝微粒体的代谢稳定性测定表明,与 相比,氘代衍生物的半衰期显着增加。在异种移植小鼠模型中,1 在 40 mg/kg 剂量下抑制肿瘤生长,抑制率为 75.1%,而 TGI 与其非氘化化合物在 80 mg/kg 剂量下的抑制率为 73.2%。机制研究表明,它是一种有效的 EGFR 和 EGFR 激酶抑制剂,可下调 EGFR 和 m-TOR 信号通路的蛋白磷酸化,通过影响 CDC25C 的表达将细胞周期阻滞在 G2/M 期,并通过调节 CDC25C 的表达来促进细胞凋亡。切割的 caspase-3 的表达。总之,可以作为一种有前途的氘化化合物,用于开发高效抗癌剂。






























 京公网安备 11010802027423号
京公网安备 11010802027423号