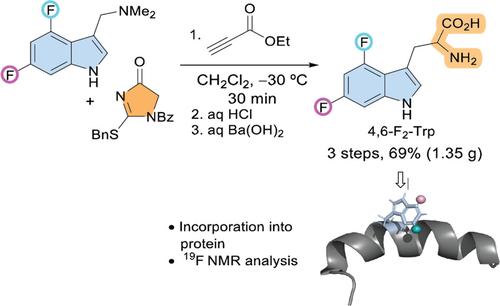
使用氟化氨基酸类似物将单个氟原子以尽可能多的链位置选择性掺入蛋白质中,是评估蛋白质结构和配体结合的一种有吸引力的方法。在这方面最常用的氟化氨基酸之一是色氨酸(Trp)。单氟化色氨酸可以很容易地掺入蛋白质中,而不需要专门的菌株。 7除了单氟化色氨酸之外,我们的目标是将这一研究空间扩大到二氟色氨酸,提供不止一种探针来评估氟化蛋白质的行为。这就需要方便且容易地获得此类氟化氨基酸。在这里,我们报告了 4,6-二氟色氨酸的便捷且可扩展的合成、将其掺入蛋白质以及这种定制氨基酸作为19 F NMR 探针的表征(方案 1)。
方案一
在图查看器PowerPoint中打开
这项工作的概念化。
为了合成目标氟化色氨酸1 ,我们的计划设想将相应的氟化禾胺与亲核甘氨酸等价物偶联。禾本科胺(3-氨基甲基吲哚)在各种活化机制下往往会失去连接在苄基位置的 R 1 R 2 N 单元,生成亲电子的氮杂富烯中间体,该中间体可以被多种亲核试剂原位拦截。 8特别是,Sainsbury 9报道称,在平稳条件下,作为二甲氨基清除剂的二甲基乙炔二羧酸酯可以促进禾本科胺分别与丙二腈或 α-氰基酯的偶联。 Williams 对该方法进行了进一步改进,确定丙炔酸乙酯3是最佳活化试剂10 (方案 2)。然而,据报道,在 Williams 条件下(Et 2 O 作为溶剂,室温 15 分钟),禾本科胺2a与N- (二苯基亚甲基)甘氨酸甲酯6a偶联,以低 41% 的分离率提供色氨酸衍生物7a。屈服。从一开始,考虑到这些先例,该方法生产所需氟化色氨酸的适用性尚不清楚。
方案2
在图查看器PowerPoint中打开
丙炔酸乙酯促进了禾本科胺2a与甘氨酸等价物的偶联。 (Williams等人,参见参考文献10 )。
为了对该方法进行初步评估,二氟化禾胺2b (通过曼尼希反应从相应的吲哚11合成)与亚氨基酯6a在稍微过量(1.1当量)的丙炔酸乙酯3存在下在报道的最佳条件下进行反应由威廉姆斯在 0 °C 时进行。确实获得了所得加合物7b ,尽管毫不奇怪,分离产率较低(22%)。将溶剂从 Et 2 O 改为 CH 2 Cl 2将产率提高至但仍不令人满意的 29%(方案 3a)。
方案3
在图查看器PowerPoint中打开
亚氨基酯6a和 2-苄基硫代咪唑酮6b与二氟禾本科胺2b偶联的比较。
在可能容易修改的其他反应参数中,我们重点关注除 α-亚氨基酯之外的合理甘氨酸等价物。在这方面,最近我们中的一些人引入了 2-苄基硫代咪唑啉酮作为亲核乙内酰脲替代物,其在(弱碱性)有机催化条件下顺利添加到各种迈克尔受体上。 12在此基础上,我们推断,将 2-苄基硫代咪唑酮6 b的 C-4 亲核特性与原位形成的亲电子氮杂富烯(如5 b )配对可能是有利的,为上述问题提供了解决方案,因为乙内酰脲随后可以转化为各自的α-氨基酸。 13为此,令人欣慰的是观察到6 b与2 b在 CH 2 Cl 2溶剂中于 0 °C 反应,得到偶联加合物8 ,分离产率有望达到 54%(方案 3b)。进一步的实验表明,温度对反应结果影响很大:在 -35 °C 下进行反应导致形成分析纯材料化合物8 ,分离收率为 95%。具有实际重要性的是,可以将反应规模扩大到数克规模,而不会对产物收率或纯度造成任何显着损害,从而可以从 1.25 g (5.94 mmol) 禾本科胺2 b开始制备 2.54 g (5.34 mmol) 二氟化加合物8 ( 90% 的产率)在一次运行中。
在0℃(冰浴)下,向乙内酰脲9 (1当量,3.70mmol,1.37g)在35mL H 2 O中的溶液中加入Ba(OH) 2一水合物(4当量,14.8mmol,2.80g)额外。将所得混合物回流96小时,观察到氨气的放出和碳酸钡的形成。然后将混合物冷却至室温,过滤沉淀并用水洗涤。将水层用HCl 3M (6当量,22.2mmol,3.7mL)酸化,然后首先在旋转蒸发器中减压除去水和挥发物(HCl),最后在真空泵下除去。将所得固体溶解在水中,然后装入 dowex® 50WX2 氢型(50–100 目)离子交换树脂(预先用 1.0 M HCl 清洗)。然后,用水处理树脂直至洗脱液的pH为中性。然后,通过5%NH 3水溶液洗脱使化合物从树脂中释放。将获得的碱性溶液真空浓缩,得到两性离子形式的 4,6-二氟-Trp 1 ,为浅棕色固体,mp=259–262 °C。产量:0.83克,3.47毫摩尔,94%。 1H NMR (300 MHz , D 2 O)δ =7.22 (s, 1H), 7.08 (dd, J HF =9.6, J HH =2.1 Hz, 1H), 6.83–6.69 (m, 1H), 4.01 (dd , J = 8.9,4.9 Hz,1H),3.53(dd, J = 15.1,4.8 Hz,1H),3.20(dd, J = 15.0,9.0 Hz,1H)。 13C{ 1 H} NMR(126MHz , CD 3 OD)δ =173.9,160.6(dd, JCF= 237.3,12.3Hz),157。7(dd, JCF = 246.0,15.3 Hz),140.3(t, JCF = 14.7 Hz),126.0(d, JCF = 2.9 Hz),113.8(d, JCF = 19.4 Hz),108.9,95.4( dd, JCF = 29.3,24.4 Hz),94.9(dd, JCF = 26.0,4.4 Hz),57.3,29.3。 19F NMR(376MHz , CD 3 OD )δ-121.54(td, JFH/FF =9.9,4.1Hz),-122.51(dd, JFH/FF =11.2,4.3Hz)。 HRMS (ESI)m / z :[M+H] + C 11 H 11 F 2 N 2 O 2的计算值,241.0783;发现 241.0789。 IR (cm -1 ):3457、1646、1557、1540、1507。
蛋白质制备
如前所述,用编码HIV衣壳-CTD的构建体的质粒转化BL21(DE3)大肠杆菌。 15培养物在 37°C 下在含有 100 μg/L 羧苄青霉素、1 g/L− 15 NH 4 Cl 和 2 g/L 葡萄糖分别作为唯一氮源和碳源的 M9 改良基本培养基中生长。在 OD 600为 0.6–0.7 时,向培养物中添加 1 g/L 草甘膦和 100 mg/L 两种芳香族氨基酸(苯丙氨酸和酪氨酸)以及 50 mg/L 氟化氨基酸 4-氟色氨酸、6-氟色氨酸或4,6-二氟色氨酸。 45 分钟后,用 1 mM IPTG 诱导蛋白表达。然后将培养物在 18°C 下再生长 16 小时。 4°C、3,500 g 离心 30 分钟收获细胞。将细胞沉淀重悬于 40 mL 裂解缓冲液(25 mM 磷酸钠,pH 5.8,5 mM DTT)中,并通过超声处理裂解。 4°C、33,000 g 离心 40 分钟去除细胞碎片。将澄清的裂解物加载到 5 mL 离子交换柱(HiTrap SP HP,5 mL)上,并使用 25 mM 磷酸钠缓冲液(pH 5.8、5 mM DTT)中的 0–50% 1 mM NaCl 梯度进行洗脱。收集、合并 10-15% NaCl 之间洗脱的蛋白质和含有蛋白质的级分,并在 Amicon 中以 3000 Da 截止值浓缩。使用含有 2 mM DTT 的磷酸盐缓冲盐水(pH 7.4),通过 Superdex 75 10/300 GL 柱(GE Healthcare Life Sciences)通过尺寸排阻对浓缩的蛋白质进行分级。通过 PAGE 和质谱法评估蛋白质纯度和 F 掺入。
19 F核磁共振波谱
在 14.1 T Bruker AVANCE 光谱仪上,在 298 K 下记录同一水缓冲液中单个氨基酸和氟化蛋白质的所有19 F 光谱,该光谱仪配备 CP TXO F/CHD 三重共振、z 轴梯度冷冻探针,温度为 283 K。 19 F化学位移参考三氟乙酸。使用 5 秒的循环延迟收集19 F 光谱,包含 16,000 个数据点和 50 ppm 的光谱宽度。载波频率设置为-123 ppm。所有光谱均使用 Bruker Topspin 3.2 进行处理。化合物1 、 2b 、 7b 、 8和9的19 F 光谱是在配备有 Iprobe 的 Bruker Avance Neo 400 上于 298 K 下以 376.5 MHz 记录,使用单氟苯作为内参比。这些光谱使用 Topspin 4.1.4 和 Mestrenova 软件进行处理。
致谢
我们感谢巴斯克政府(EJ,赠款 IT1583-22)和 Agencia Estatal de Investigación(赠款 PID2019-109633GB-C21/AEI/10.13039/501100011033 和 PID2022-137153NB-C21/AEI/10.13039/501100011033 )以获得财政支持。作者感谢 SGIker(UPV/EHU/ERDF、EU)提供的技术和人力支持。 IH 感谢 EJ 的奖学金。 RM-P。得到了匹兹堡大学健康科学多样性学者计划和美国心脏协会博士后奖学金的支持。格罗南博恩实验室的工作得到了 NIH 拨款 U54AI170791 的支持。
"点击查看英文标题和摘要"
Synthesis of 4,6-Difluoro-Tryptophan as a Probe for Protein 19F NMR
Given the increasingly growing number of new pharmaceuticals containing fluorine atoms,1 fluorine chemistry and applications of fluorinated compounds are becoming a flourishing areas of research. Fluorinated amino acids are in high demand,2 and their synthesis3 and incorporation into proteins are currently a very exciting and expanding subjects. Nowadays, application of fluorinated tailor-made amino acids to study protein dynamics, structure, folding and function is well beyond drug discovery campaigns, with fluorine NMR spectroscopy gaining much popularity as a valuable tool.4 The 100% naturally abundant 19F atom possesses favourable NMR properties: a spin 1/2 nucleus and the second highest gyromagnetic ratio after hydrogen, resulting in excellent sensitivity.5 In addition, the resonance frequency of the fluorine nucleus is exquisitely responsive to its conformational and electronic environment, covering a range of >300 ppm.6 Furthermore, since 19F is virtually absent from all biological molecules, spectra of fluorinated proteins do not suffer from any background signals.
Incorporation of single fluorine atoms into proteins with as much chain position selectivity as possible, using fluorinated amino acid analogues, is an attractive approach to assess protein structure as well as ligand binding. One of the most used fluorinated amino acids in that regard is tryptophan (Trp). Monofluorinated tryptophans can be readily incorporated into proteins without the need of specialized strains.7 Beyond monofluorinated tryptophans, here we aimed to enlarge this research space to difluoro-tryptophans, providing more than one probe to assess the behavior of the fluorinated protein. This necessitates convenient and facile access to such fluorinated amino acids. Here, we report an expedient and scalable synthesis of 4,6-difluoro-tryptophan, its incorporation into a protein and the characterization of this tailor-made amino acid as a probe in 19F NMR (Scheme 1).
Scheme 1
Open in figure viewerPowerPoint
Conceptualization of this work.
For the synthesis of the target fluorinated tryptophan 1, our plan envisioned the coupling of the corresponding fluorinated gramine with a nucleophilic glycine equivalent. Gramines (3-aminomethylindoles), tend to lose the R1R2N unit attached at the benzylic position under various activation mechanisms, generating an electrophilic azafulvene intermediate which can be intercepted with a variety of nucleophiles in situ.8 In particular, Sainsbury9 reported that coupling of gramines with malononitrile or an α-cyanoester, respectively, could be promoted by dimethylacetylene dicarboxylate, which serves as the dimethylamino group scavenger, under smooth conditions. Further refinement of the method by Williams identified ethyl propiolate 3 as the optimal activating reagent10 (Scheme 2). However, the coupling of gramine 2 a with N-(diphenylmethylene)glycine methyl ester 6 a under Williams conditions (Et2O as solvent, 15 min at room temperature) was reported to provide the tryptophan derivative 7 a in a low 41% isolated yield. At the outset, and in view of these precedents, the suitability of the approach for the production of the desired fluorinated tryptophan was unclear.
Scheme 2
Open in figure viewerPowerPoint
Coupling of gramine 2 a with a glycine equivalent promoted by ethyl propiolate. (Williams et al. in ref 10).
For initial assessment of the approach, difluorinated gramine 2 b (synthesized through the Mannich reaction from the corresponding indole11) was subjected to reaction with iminoester 6 a in the presence of slight excess (1.1 equivalents) of ethyl propiolate 3 under the optimal conditions reported by Williams at 0 °C. The resulting adduct 7 b was indeed obtained, although, not surprisingly, in a low isolated yield (22%). Changing the solvent from Et2O to CH2Cl2 improved the yield to a yet unsatisfactory 29% (Scheme 3a).
Scheme 3
Open in figure viewerPowerPoint
Comparison of iminoester 6 a and 2-benzylthioimidazolone 6 b in the coupling with difluorogramine 2 b.
Among the additional reaction parameters that might be susceptible for modification, we focused on plausible glycine equivalents other than α-iminoesters. In this respect, recently some of us introduced 2-benzylthioimidazolones as pronucleophilic hydantoin surrogates that add smoothly under (slightly basic) organocatalytic conditions to various Michael acceptors.12 On this basis, we reasoned that pairing the C-4 pronucleophilic character of 2-benzylthioimidazolone 6 b with in situ formed electrophilic azafulvenes, like 5 b, might be favorable, providing a solution to the above problem since hydantoins may afterwards be transformed into the respective α-amino acid.13 To that end, it was gratifying to observe that 6 b reacted with 2 b, in CH2Cl2 solvent, at 0 °C to afford the coupling adduct 8 in a promising 54% isolated yield (Scheme 3b). Further experimentation revealed that the temperature influenced the reaction outcome dramatically: running the reaction at −35 °C resulted in formation of compound 8 as analytically pure material in 95% isolated yield. Of practical importance, scaling up the reaction to the multigram scale was possible without any significant detriment on product yield or purity, allowing preparation of 2.54 g (5.34 mmol) of difluorinated adduct 8 starting from 1.25 g (5.94 mmol) of gramine 2 b (90% yield) in a single run.
Treatment of 8 with 6 M HCl in dioxane at room temperature for 16 h gave rise to the corresponding N-benzoyl hydantoin 9. Then, straightforward hydantoin ring opening through basic hydrolysis,14 protonation and purification under an ion-exchange column, yielded the target difluorinated amino acid 1, with an overall 65% yield (three steps, Scheme 4).
Scheme 4
Open in figure viewerPowerPoint
Hydrolytic conversion of coupling adduct 8 to hydantoin 9 and target 4,6-F2-Trp 1.
All three fluorinated tryptophans, 4-fluoro tryptophan, 6-fluoro tryptophan and the synthesized 4,6-difluoro tryptophan were efficiently incorporated into protein, and yields of labeled protein were essentially identical. This indicates that the 4,6-difluoro tryptophan is efficiently used by the biosynthetic protein synthesis machinery of E. coli. Note, in contrast to perfluorinated compounds, the two fluorine atoms on one amino acid in the CTD do not change the property of the protein significantly, although the dimerization constants seem to have been affected (see SI).
This domain dimerizes (Figure 1), and the dimer links two adjacent hexameric units in the overall capsid lattice via an antiparallel helix-helix interface. At the center of the interface, the single Trp residue in this domain plays a pivotal role in the stabilization of the extended lattice of the conical capsid shell.15 As can be appreciated, both the 4- and 6-position of the six membered indole ring are separated by 5.2 Å (4F–4F), 6.1 Å (6F–6F) and 5.4 Å across the dimer interface in the structure, ideally suited to report on dimer formation. The WT CTD forms a dimer with a Kd value of ∼10 μM. Interestingly, two different dimers were observed for WT CTD as well as 4-fluoro-Trp CTD, labeled D1 and D2 in Figure 1, whose relative amounts are both pH, temperature and concentration dependent.16 Given the crucial positions of the fluorine atoms in 4-fluoro-Trp CTD, 6-fluoro-Trp CTD and 4,6-difluoro-Trp CTD, we used 19F NMR to evaluate the protein. Spectra of both the free fluorinated amino acid and after incorporation into CA-CTD are displayed in Figure 1. Clearly, distinct 19F chemical shifts are seen, with the 4-F and 6-F resonances of the isolated amino acids located at −46.4 ppm and −43.7 ppm, respectively. Interestingly, the equivalent resonances in 4,6-difluoro-Trp are located at −41.5 and −43.5 ppm, distinctly different from the resonance positions of the singly fluorinate Trp amino acids. As was noted previously, the isotropic 19F chemical shift of a fluorine atom at the 6 position in Trp is downfield of that at the 4 position,17 also seen here. In the 4-F-Trp CA-CTD protein spectra, both dimer forms (D1 and D2) are seen, as well as the monomer (M), while for 6-F-Trp CA-CTD only in the 10 μM sample the resonance for the monomer can be detected. Interestingly, however, the 19F spectrum of the 4,6-F2-Trp CTD shows four, not well-resolved resonances. Recording spectra at different concentrations (SI) did not permit unambiguous assignment of the resonances to the monomer and dimer species, different from the case of the monofluorinated 4F-Trp and 6-F-Trp CTD. Gratifyingly, though, the biosynthetic incorporation of 4,6-F2-Trp into the C-terminal domain of the HIV-1 capsid protein (CA-CTD) was achieved, indicating that the E. coli protein synthesis machinery is capable of utilizing this di-fluorinated amino acid.
Figure 1
Open in figure viewerPowerPoint
HIV1 CA-CTD (PDB ID; 2KOD) in ribbon representation with the Trp side chain at the dimer interface depicted in stick representation and the F atoms at the 4 and 6 positions as cyan and magenta spheres, respectively (top left). SDS PAGE of 4-F-, 6-F- and 4,6-F Trp CA-CTD with marker sizes indicated (bottom left). 19F spectra at 25 °C of 4-F, 6-F and 4,6-F2-Trp at 10 μM protein concentration. 4-F, 6-F and 4,6-F2-Trp CA-CTD spectra are shown in cyan, magenta and blue, respectively. D, dimer; M, monomer.
In conclusion, a scalable procedure for the synthesis of 4,6-difluorotryptophan 1 (4,6-F2-Trp) is reported based on a deaminative coupling between a 4,6-difluorogramine 2 b and 2-benzylthio-1,5-dihydro-4H-imidazolone 6 b. Key for a clean reaction and efficient coupling is to keep the reaction temperature low (−35 °C) and to use 6 b as an advantageous glycine synthetic equivalent. The incorporation of prepared 4,6-F2-Trp into the C-terminal domain of the HIV-1 capsid protein (CA-CTD) was realised with similar yield as with the individual mono fluorinated amino acids.
Experimental Section
Ethyl Propiolate-Promoted Addition of 2-Benzylthioimidazolone 6 b to Gramine 2 b
To a solution of gramine 2 b (42 mg, 0.2 mmol) in dry CH2Cl2 (2 mL) at room temperature, 2-benzylthioimidazolone 6 b (2 eq., 0.4 mmol, 124 mg) was added. After cooling the solution to −40 °C, ethyl propiolate 3 (1.1 eq., 0.22 mmol, 22 mg) was added and the reaction mixture was stirred at the same temperature for 10 minutes. Afterwards, the mixture was allowed to reach −35 °C slowly over 30 minutes. Then, the solvent was evaporated under reduced pressure and the crude compound was purified by column chromatography (eluent: hexane/ethyl acetate 5:1) to obtain 8 as a brown solid, m.p.=215–217 °C. Yield 90.4 mg, 0.19 mmol, 95%. 1H NMR (300 MHz, CDCl3)δ=8.01 (s, 1H), 7.42–7.13 (m, 10H), 6.83 (d, J=2.4 Hz, 1H), 6.74 (dd, JHF=8.9, JHH=2.1 Hz, 1H), 6.50–6.39 (m, 1H), 5.13–5.04 (m, 1H), 4.36 (d, J=2.4 Hz, 2H), 3.34 (dd, J=15.3, 4.8 Hz, 1H), 3.19 (dd, J=15.1, 6.1 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3)δ=185.8, 185.1, 167.5, 159.5 (dd, J=239.8, 12.2 Hz), 156.2 (dd, J=248.1, 15.1 Hz), 137.7 (t, J=14.0 Hz), 135.2, 132.7, 132.5, 129.6, 128.9, 128.6, 128.0, 127.9, 124.1 (d, J=3.2 Hz), 112.8 (d, J=19.4 Hz), 107.2 (d, J=3.1 Hz), 95.8 (dd, J=28.7, 23.9 Hz), 93.9 (dd, J=26.1, 4.4 Hz), 65.9, 38.3, 28.5 (d, J=2.0 Hz). 19F NMR (376 MHz, CDCl3) δ −118.14 (td, JFH/FF=9.2, 4.6 Hz), −119.48 (dd, JFH/FF=11.0, 4.8 Hz). HRMS (ESI)m/z [M + H]+ Calcd for C26H20F2N3O2S, 476.1239; found 476.1246. IR (cm−1) 3354, 1717, 1674.
Large Scale Synthesis of 8
The same procedure as above was followed from 1.25 g (5.94 mmol) of starting gramine 2 b and using the following quantities of solvent and reagents: 50 mL dry CH2Cl2, 2-benzylthioimidazolone 6 b (2 eq., 11.88 mmol, 3.69 mg) and ethyl propiolate 3 (1.1 eq., 6.54 mmol, 0.64 g). Yield 2.54 g, 5.34 mmol, 90%.
Hydrolysis of 8 into Fluorinated Hydantoin 9
A solution of aq. HCl 6 M (11 eq. 58.74 mmol, 9.79 mL) was added dropwise to a solution of imidazolone 8 (5.34 mmol, 2.54 g) in 1,4-dioxane (80 mL) at 0 °C. Once the addition was complete, the reaction was stirred at room temperature overnight. Afterward, the mixture was cooled to 0 °C and a saturated solution of NaHCO3 was added until basic pH was reached. The aqueous layer was extracted with CH2Cl2 threefold and the combined organic layers were dried over MgSO4 and the solvent evaporated under reduced pressure. The crude product was purified by silica gel flash column chromatography (hexane/EtOAc 3:1 to 1:1) to obtain hydantoin 9 as a white foam. Yield: 1.37 g, 3.70 mmol, 69%. 1H NMR (300 MHz, CDCl3)δ=8.46 (s, 1H), 8.11 (s, 1H), 7.54–7.23 (m, 5H), 6.96 (d, J=2.3 Hz, 1H), 6.83 (dd, JHF=8.9, JHH=2.0 Hz, 1H), 6.43 (ddd, JHF=11.0, 9.9, JHH=2.0 Hz, 1H), 5.24 (dd, J=5.2, 3.3 Hz, 1H), 3.79 (dd, J=15.1, 5.2 Hz, 1H), 3.56–3.43 (dd, J=15.1, 3.3 Hz, 1H). 13C NMR {1H} (126 MHz, CD3OD)δ=174.4, 170.1, 160.3 (dd, JCF=237.2, 12.0 Hz), 157.4 (dd, JCF=247.3, 15.0 Hz), 155.0, 139.4 (t, JCF=14.3 Hz), 135.5, 133.0, 130.2, 128.4, 126.2 (d, JCF=2.3 Hz), 114.6 (d, JCF=18.7 Hz), 107.0 (d, JCF=2.9 Hz), 95.5 (dd, JCF=29.2, 24.0 Hz), 94.8 (dd, JCF=26.0, 4.4 Hz), 62.9, 25.9 (d, JCF=2.5 Hz). 19F NMR (376 MHz, CDCl3) δ −117.73 (td, JFH/FF=9.5, 4.5 Hz), −118.41 (dd, JFH/FF=11.1, 4.6 Hz). HRMS (ESI)m/z [M+H]+ Calcd for C19H14F2N3O3, 370.0998; found, 370.1005. IR (cm−1): 3232, 1791, 1731.
Conversion of Hydantoin 9 into Fluorinated Tryptophan 1
To a solution of hydantoin 9 (1 eq., 3.70 mmol, 1.37 g) in 35 mL H2O at 0 °C (ice bath), Ba(OH)2 monohydrate (4 eq., 14.8 mmol, 2.80 g) was added. The resulting mixture was refluxed for 96 hours, with evolution of ammonia gas and formation of barium carbonate being observed. The mixture was then cooled down to room temperature and the precipitate was filtered and washed with water. The aqueous layer was acidified with HCl 3 M (6 eq., 22.2 mmol, 3.7 mL) and then water and the volatiles (HCl) were removed under reduced pressure in the rotary evaporator first and finally under vacuum pump. The resulting solid was dissolved in water and then loaded into a dowex® 50WX2 hydrogen form (50–100 mesh) ion exchange resin (previously washed with 1.0 M HCl). Afterwards, the resin was treated with water until the pH of the eluent was neutral. Then, the compound was liberated from the resin by eluting a 5% NH3 aqueous solution. The obtained basic solution was concentrated under vacuum to yield the 4,6-difluoro-Trp 1 on its zwitterionic form as a pale brown solid, m.p.=259–262 °C. Yield: 0.83 g, 3.47 mmol, 94%. 1H NMR (300 MHz, D2O)δ=7.22 (s, 1H), 7.08 (dd, JHF=9.6, JHH=2.1 Hz, 1H), 6.83–6.69 (m, 1H), 4.01 (dd, J=8.9, 4.9 Hz, 1H), 3.53 (dd, J=15.1, 4.8 Hz, 1H), 3.20 (dd, J=15.0, 9.0 Hz, 1H). 13C{1H} NMR (126 MHz, CD3OD)δ=173.9, 160.6 (dd, JCF=237.3, 12.3 Hz), 157.7 (dd, JCF=246.0, 15.3 Hz), 140.3 (t, JCF=14.7 Hz), 126.0 (d, JCF=2.9 Hz), 113.8 (d, JCF=19.4 Hz), 108.9, 95.4 (dd, JCF=29.3, 24.4 Hz), 94.9 (dd, JCF=26.0, 4.4 Hz), 57.3, 29.3. 19F NMR (376 MHz, CD3OD) δ −121.54 (td, JFH/FF=9.9, 4.1 Hz), −122.51 (dd, JFH/FF=11.2, 4.3 Hz). HRMS (ESI)m/z: [M+H]+ Calcd for C11H11F2N2O2, 241.0783; found 241.0789. IR (cm−1): 3457, 1646, 1557, 1540, 1507.
Protein Preparation
BL21 (DE3) E. coli were transformed with a plasmid encoding a construct coding for the HIV capsid-CTD, as previously described.15 Cultures were grown at 37 °C in M9 modified minimal medium containing 100 μg/L Carbenicillin, 1 g/L−15NH4Cl, and 2 g/L of glucose as the sole nitrogen and carbon sources, respectively. At OD600 of 0.6–0.7, the culture was supplemented with 1 g/L of glyphosate and 100 mg/L of the two aromatic amino acids, phenylalanine and tyrosine as well as 50 mg/L of the fluorinated amino acid, 4-fluoro tryptophan, 6-fluoro tryptophan or 4,6-difluoro tryptophan. After 45 minutes, protein expression was induced with 1 mM IPTG. The culture was then grown at 18 °C for an additional 16 h. Cells were harvested by centrifugation at 3,500 g for 30 min at 4 °C. Cell pellets were resuspended in 40 mL lysis buffer (25 mM sodium phosphate, pH 5.8, 5 mM DTT) and lysed by sonication. Cell debris was removed by centrifugation at 33,000 g for 40 min at 4 °C. The clarified lysate was loaded onto 5 mL ion-exchange column (HiTrap SP HP, 5 mL) and eluted using a gradient from 0–50% 1 mM NaCl in 25 mM sodium phosphate buffer, pH 5.8, 5 mM DTT. Protein eluted between 10–15% NaCl and protein containing fractions were collected, pooled, and concentrated in an Amicon with a 3000 Da cut-off. The concentrated protein was fractionated by size-exclusion over a Superdex 75 10/300 GL column (GE Healthcare Life Sciences) using phosphate buffered saline, pH 7.4, containing 2 mM DTT. Protein purity and F incorporation was assessed by PAGE and mass spectrometry.
19F NMR Spectroscopy
All 19F spectra of individual amino acids and fluorinated proteins in the same aqueous buffer were recorded at 298 K on a 14.1 T Bruker AVANCE spectrometer, equipped with a CP TXO F/C-H-D triple-resonance, z-axis gradient cryoprobe at 283 K. 19F chemical shifts were referenced to trifluoracetic acid. 19F spectra were collected with 16,000 data points and a spectral width of 50 ppm using a recycle delay of 5 s. The carrier frequency was set to −123 ppm. All spectra were processed using Bruker Topspin 3.2. 19F spectra of compounds 1, 2 b, 7 b, 8 and 9 were recorded at 298 K on a Bruker Avance Neo 400, equipped with Iprobe, at 376.5 MHz using monofluorobenzene as internal reference. These spectra were processed using Topspin 4.1.4 and Mestrenova software.
Acknowledgments
We thank the Basque Government (EJ, grant IT1583-22) and Agencia Estatal de Investigación (grants PID2019-109633GB-C21/AEI/10.13039/501100011033 and PID2022-137153NB-C21/AEI/10.13039/501100011033) for financial support. The authors are grateful for the technical and human support provided by SGIker (UPV/EHU/ERDF, EU). I.H. thanks EJ for a fellowship. R.M-P. was supported by the Health Sciences Diversity Scholars Program at the University of Pittsburgh and F.B. by a post-doctoral fellowship from the American Heart Association. The work in the Gronenborn laboratory was supported by NIH grant U54AI170791.


































 京公网安备 11010802027423号
京公网安备 11010802027423号