当前位置:
X-MOL 学术
›
Mol. Genet. Genomic Med.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Okur‐Chung neurodevelopmental syndrome: Implications for phenotype and genotype expansion
Molecular Genetics & Genomic Medicine ( IF 1.5 ) Pub Date : 2024-03-06 , DOI: 10.1002/mgg3.2398 Haitian Nan 1 , Min Chu 1 , Jing Zhang 1 , Deming Jiang 1 , Yihao Wang 1 , Liyong Wu 1, 2
Molecular Genetics & Genomic Medicine ( IF 1.5 ) Pub Date : 2024-03-06 , DOI: 10.1002/mgg3.2398 Haitian Nan 1 , Min Chu 1 , Jing Zhang 1 , Deming Jiang 1 , Yihao Wang 1 , Liyong Wu 1, 2
Affiliation
|
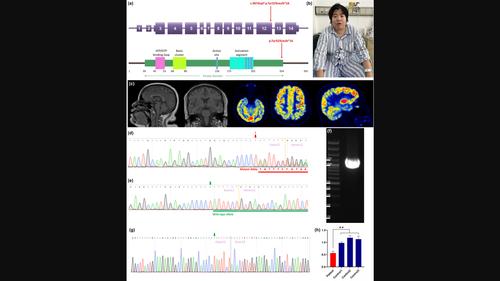
BackgroundOkur‐Chung neurodevelopmental syndrome (OCNDS) is a rare autosomal dominant disorder caused by pathogenic variants in CSNK2A1 . It is characterized by intellectual disability, developmental delay, and multisystemic abnormalities.MethodsWe performed the whole‐exome sequencing for a patient in a Chinese family. The co‐segregation study using the Sanger sequencing method was performed among family members. Reverse transcription and quantitative real‐time polymerase chain reaction were carried out using total RNA from blood samples of the proband and wild‐type control subjects. A review of patients with OCNDS harboring CSNK2A1 pathogenic variants was conducted through a comprehensive search of the PubMed database.ResultsWe identified a novel CSNK2A1 frameshift variant p.Tyr323Leufs*16 in a Chinese family. The proband, a 31‐year‐old female, presented with abnormal eating habits, recurrent seizures, language impairment, and intellectual disability. Her mother exhibited postnatal hernias, splenomegaly, and a predisposition to infections, but showed no significant developmental impairments or intellectual disability. Genetic studies revealed the presence of this variant in CSNK2A1 in both the proband and her mother. Transcription analysis revealed this variant may lead to nonsense‐mediated mRNA decay, suggesting haploinsufficiency as a potential disease mechanism. We reviewed 47 previously reported OCNDS cases and discovered that individuals carrying CSNK2A1 null variants may exhibit a diminished frequency of symptoms linked to language deficits, dysmorphic facial features, or intellectual disability, consequently presenting an overall milder phenotype when compared to those with missense variants.ConclusionWe report a novel frameshift variant, p.Tyr323Leufs*16, in an OCNDS family with a generally mild phenotype. This study may broaden the spectrum of clinical presentations associated with OCNDS and contribute novel insights into the genotype–phenotype correlation of this condition.
更新日期:2024-03-06





























 京公网安备 11010802027423号
京公网安备 11010802027423号