当前位置:
X-MOL 学术
›
Redox Biol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
IL-4 activates ULK1/Atg9a/Rab9 in asthma, NLRP3 inflammasomes, and Golgi fragmentation by increasing autophagy flux and mitochondrial oxidative stress
Redox Biology ( IF 10.7 ) Pub Date : 2024-02-15 , DOI: 10.1016/j.redox.2024.103090 Chang Xu 1 , Yilan Song 1 , Wanting Liu 1 , Ruobai Liu 1 , Qiaoyun Bai 1 , Liangchang Li 1 , Chongyang Wang 1 , Guanghai Yan 1
Redox Biology ( IF 10.7 ) Pub Date : 2024-02-15 , DOI: 10.1016/j.redox.2024.103090 Chang Xu 1 , Yilan Song 1 , Wanting Liu 1 , Ruobai Liu 1 , Qiaoyun Bai 1 , Liangchang Li 1 , Chongyang Wang 1 , Guanghai Yan 1
Affiliation
|
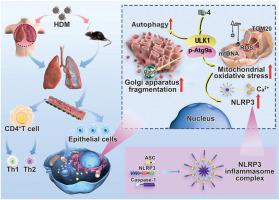
During asthma, there is an intensification of pulmonary epithelial inflammation, mitochondrial oxidative stress, and Golgi apparatus fragmentation. However, the underlying mechanism remains largely unknown. Therefore, this study investigated the roles of ULK1, Atg9a, and Rab9 in epithelial inflammation, mitochondrial oxidative stress, and Golgi apparatus fragmentation. We found that ULK1 gene knockout reduced the infiltration of inflammatory cells, restored the imbalance of the Th1/Th2 ratio, and inhibited the formation of inflammatory bodies in the lung tissue of house dust mite-induced asthma mice. Moreover, we demonstrated that Atg9a interacted with ULK1 at S467. ULK1 phosphorylated Atg9a at S14. Treatment with ULK1 activator (LYN-1604) and ULK1 inhibitor (ULK-101) respectively promoted and inhibited inflammasome activation, indicating that the activation of inflammasome induced by house dust mite in asthma mice is dependent on ULK1. For validation of the results, we then used a lentivirus containing ULK1 wild type and ULK1-S467A genes to infect Beas-2b-ULK1-knockout cells and establish a stable cell line. The results suggest that the ULK1 S467 site is crucial for IL-4-induced inflammation and oxidative stress. Experimental verification confirmed that Atg9a was the superior signaling pathway of Rab9. Interestingly, we found for the first time that Rab9 played a very important role in inflammation-induced fragmentation of the Golgi apparatus. Inhibiting the activation of the ULK1/Atg9a/Rab9 signaling pathways can inhibit Golgi apparatus fragmentation and mitochondrial oxidative stress in asthma while reducing the production of NLRP3-mediated pulmonary epithelial inflammation.
中文翻译:

IL-4 通过增加自噬通量和线粒体氧化应激来激活哮喘、NLRP3 炎症小体和高尔基体断裂中的 ULK1/Atg9a/Rab9
哮喘期间,肺上皮炎症、线粒体氧化应激和高尔基体破碎加剧。然而,其根本机制仍然很大程度上未知。因此,本研究调查了 ULK1、Atg9a 和 Rab9 在上皮炎症、线粒体氧化应激和高尔基体断裂中的作用。我们发现ULK1基因敲除减少了炎症细胞的浸润,恢复了Th1/Th2比例的不平衡,并抑制了屋尘螨诱发的哮喘小鼠肺组织中炎症小体的形成。此外,我们证明 Atg9a 在 S467 处与 ULK1 相互作用。 ULK1 在 S14 处磷酸化 Atg9a。 ULK1激活剂(LYN-1604)和ULK1抑制剂(ULK-101)治疗分别促进和抑制炎症小体激活,表明屋尘螨诱导哮喘小鼠炎症小体激活依赖于ULK1。为了验证结果,我们使用含有 ULK1 野生型和 ULK1-S467A 基因的慢病毒感染 Beas-2b-ULK1 敲除细胞并建立稳定的细胞系。结果表明 ULK1 S467 位点对于 IL-4 诱导的炎症和氧化应激至关重要。实验验证证实Atg9a是Rab9的优势信号通路。有趣的是,我们首次发现Rab9在炎症引起的高尔基体碎裂中发挥着非常重要的作用。抑制 ULK1/Atg9a/Rab9 信号通路的激活可以抑制哮喘中的高尔基体断裂和线粒体氧化应激,同时减少 NLRP3 介导的肺上皮炎症的产生。
更新日期:2024-02-15
中文翻译:
IL-4 通过增加自噬通量和线粒体氧化应激来激活哮喘、NLRP3 炎症小体和高尔基体断裂中的 ULK1/Atg9a/Rab9
哮喘期间,肺上皮炎症、线粒体氧化应激和高尔基体破碎加剧。然而,其根本机制仍然很大程度上未知。因此,本研究调查了 ULK1、Atg9a 和 Rab9 在上皮炎症、线粒体氧化应激和高尔基体断裂中的作用。我们发现ULK1基因敲除减少了炎症细胞的浸润,恢复了Th1/Th2比例的不平衡,并抑制了屋尘螨诱发的哮喘小鼠肺组织中炎症小体的形成。此外,我们证明 Atg9a 在 S467 处与 ULK1 相互作用。 ULK1 在 S14 处磷酸化 Atg9a。 ULK1激活剂(LYN-1604)和ULK1抑制剂(ULK-101)治疗分别促进和抑制炎症小体激活,表明屋尘螨诱导哮喘小鼠炎症小体激活依赖于ULK1。为了验证结果,我们使用含有 ULK1 野生型和 ULK1-S467A 基因的慢病毒感染 Beas-2b-ULK1 敲除细胞并建立稳定的细胞系。结果表明 ULK1 S467 位点对于 IL-4 诱导的炎症和氧化应激至关重要。实验验证证实Atg9a是Rab9的优势信号通路。有趣的是,我们首次发现Rab9在炎症引起的高尔基体碎裂中发挥着非常重要的作用。抑制 ULK1/Atg9a/Rab9 信号通路的激活可以抑制哮喘中的高尔基体断裂和线粒体氧化应激,同时减少 NLRP3 介导的肺上皮炎症的产生。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号