Structural Chemistry ( IF 2.1 ) Pub Date : 2024-02-13 , DOI: 10.1007/s11224-024-02278-5 Khalaf A. Jasim , Nazk Mohammed Aziz , Muhammad Ashfaq , Reza Behjatmanesh-Ardakani , Ahmed S. Faihan , Muhammad Nawaz Tahir , Ahmed S. Al-Janabi , Necmi Dege , Andre J. Gesquiere
|
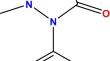
In the current work, we report the synthesis and characterization of N-((1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)carbamothioyl)benzamide by reacting 4-aminoantipyrine and benzoylisothiocynate in equimolar ratio. Moreover, the compound was characterized by single crystal XRD analysis. The various intermolecular interactions stabilized the supramolecular assembly, including H-bonding and interaction involving π-ring. Hirshfeld surface analysis was performed in order to probe intermolecular interactions in detail. Interaction energy calculations were conducted to find the type of interaction energy prominent in stabilizing supramolecular assembly. The quantum parameters of the prepared compound were investigated by utilizing the Def2-SVPD basis set in conjunction with the hybrid method of B3LYP. The results revealed quite similarities between the experimental and theoretical calculations. In addition, the HOMO orbitals are located at the hetero atoms, while the LUMO orbitals are located at the benzene ring. In addition, the prepared compound was docked with Ampicillin-CTX-M-15. The results showed good binding interaction between the ligand and the targeted amino acids, with the best binding score of − 5.26 kcal/mol.
中文翻译:
N-((1,5-二甲基-3-氧代-2-苯基-2,3-二氢-1H-吡唑-4-基)硫甲酰基)苯甲酰胺的对接、DFT和结构研究
在目前的工作中,我们报道了 N-((1,5-二甲基-3-氧代-2-苯基-2,3-二氢-1H-吡唑-4-基)硫代氨基甲酰基)苯甲酰胺的合成和表征,通过将 4等摩尔比的氨基安替比林和苯甲酰异硫氰酸酯。此外,通过单晶XRD分析对该化合物进行了表征。各种分子间相互作用稳定了超分子组装,包括氢键和涉及π环的相互作用。进行赫什菲尔德表面分析是为了详细探究分子间相互作用。进行相互作用能计算,以找出在稳定超分子组装中起主导作用的相互作用能类型。利用Def2-SVPD基组结合B3LYP的混合方法研究了所制备化合物的量子参数。结果显示实验和理论计算之间非常相似。此外,HOMO轨道位于杂原子处,而LUMO轨道位于苯环处。此外,将制备的化合物与Ampicillin-CTX-M-15对接。结果显示配体与目标氨基酸之间具有良好的结合相互作用,最佳结合得分为- 5.26 kcal/mol。





























 京公网安备 11010802027423号
京公网安备 11010802027423号