Nature Methods ( IF 36.1 ) Pub Date : 2024-02-07 , DOI: 10.1038/s41592-024-02174-0 Ben Shor 1 , Dina Schneidman-Duhovny 1
|
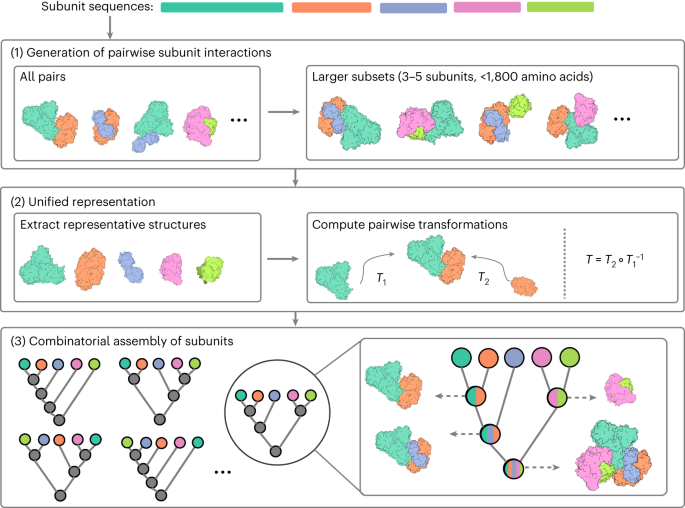
Deep learning models, such as AlphaFold2 and RosettaFold, enable high-accuracy protein structure prediction. However, large protein complexes are still challenging to predict due to their size and the complexity of interactions between multiple subunits. Here we present CombFold, a combinatorial and hierarchical assembly algorithm for predicting structures of large protein complexes utilizing pairwise interactions between subunits predicted by AlphaFold2. CombFold accurately predicted (TM-score >0.7) 72% of the complexes among the top-10 predictions in two datasets of 60 large, asymmetric assemblies. Moreover, the structural coverage of predicted complexes was 20% higher compared to corresponding Protein Data Bank entries. We applied the method on complexes from Complex Portal with known stoichiometry but without known structure and obtained high-confidence predictions. CombFold supports the integration of distance restraints based on crosslinking mass spectrometry and fast enumeration of possible complex stoichiometries. CombFold’s high accuracy makes it a promising tool for expanding structural coverage beyond monomeric proteins.
中文翻译:
CombFold:使用组合组装算法和 AlphaFold2 预测大型蛋白质组装体的结构
深度学习模型(如 AlphaFold2 和 RosettaFold)可实现高精度的蛋白质结构预测。然而,由于其大小和多个亚基之间相互作用的复杂性,大型蛋白质复合物仍然难以预测。在这里,我们介绍了 CombFold,这是一种组合和分层组装算法,用于利用 AlphaFold2 预测的亚基之间的成对相互作用来预测大蛋白质复合物的结构。CombFold 准确预测了(TM 分数 >0.7)在两个包含 60 个大型不对称组装体的数据集中排名前 10 位的复合物中的 72%。此外,与相应的蛋白质数据库条目相比,预测复合物的结构覆盖率高出 20%。我们将该方法应用于来自具有已知化学计量学但没有已知结构的 Complex Portal 的复合物,并获得了高置信度的预测。CombFold 支持基于交联质谱法的距离约束和可能的复杂化学计量的快速计数。CombFold 的高精度使其成为将结构覆盖扩展到单体蛋白之外的有前途的工具。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号