当前位置:
X-MOL 学术
›
Redox Biol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hydrogen sulfide protects cardiomyocytes from doxorubicin-induced ferroptosis through the SLC7A11/GSH/GPx4 pathway by Keap1 S-sulfhydration and Nrf2 activation
Redox Biology ( IF 10.7 ) Pub Date : 2024-01-29 , DOI: 10.1016/j.redox.2024.103066
Hui Zhang 1 , Jianan Pan 2 , Shuying Huang 2 , Xiaonan Chen 2 , Alex Chia Yu Chang 3 , Changqian Wang 2 , Junfeng Zhang 2 , Huili Zhang 2
Redox Biology ( IF 10.7 ) Pub Date : 2024-01-29 , DOI: 10.1016/j.redox.2024.103066
Hui Zhang 1 , Jianan Pan 2 , Shuying Huang 2 , Xiaonan Chen 2 , Alex Chia Yu Chang 3 , Changqian Wang 2 , Junfeng Zhang 2 , Huili Zhang 2
Affiliation
|
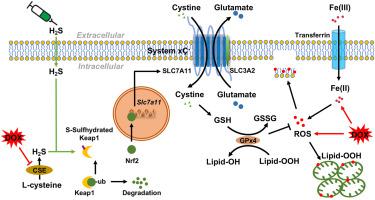
Recent studies have demonstrated that ferroptosis, a novel form of nonapoptotic regulated cell death plays an important role in doxorubicin (DOX)-induced cardiotoxicity (DoIC). Hydrogen sulfide (HS) is emerging as the third important gaseous mediator in cardiovascular system. However, whether HS has an effect on DOX-induced ferroptosis remains unknown. Here, we found that DOX not only triggered cardiomyocyte ferroptosis but also significantly inhibited the synthesis of endogenous HS in the murine model of chronic DoIC. Application of NaHS, an HS donor obviously activated the SLC7A11/GSH/GPx4 antioxidant pathway and thus alleviated DOX-induced ferroptosis and cardiac injury in mice. In contrast, cardiac-specific knockout of cystathionine γ-lyase gene () in mice (/Cre) to abolish the cardiac synthesis of endogenous HS evidently exacerbated DOX-induced ferroptosis and cardiac dysfunction. A further suppression of SLC7A11/GSH/GPx4 pathway was obtained in /Cre mice with DoIC, as compared to /Cre mice with DoIC. The aggravation caused by cardiac-specific deficiency was remarkably rescued by exogenous supplementation of NaHS. Moreover, in DOX-stimulated H9c2 cardiomyocytes, pretreatment with NaHS dose-dependently enhanced the activity of SLC7A11/GSH/GPx4 pathway and subsequently mitigated ferroptosis and mitochondrial impairment. On the contrary, transfection with siRNA in DOX-stimulated H9c2 cardiomyocytes markedly inhibited SLC7A11/GSH/GPx4 pathway, thus leading to aggravated ferroptosis and more damage to mitochondrial structure and function. In addition, the protective effect of NaHS on DOX-induced ferroptosis was closely related to the S-sulfhydrated Keap1, which in turn promoted nuclear translocation of Nrf2 and the transcription of SLC7A11 and GPx4. In conclusion, our findings suggest that HS may exert protective effect on DoIC by inhibiting DOX-induced ferroptosis via Keap1/Nrf2-dependent SLC7A11/GSH/GPx4 antioxidant pathway.
中文翻译:

硫化氢通过 Keap1 S-硫化和 Nrf2 激活通过 SLC7A11/GSH/GPx4 途径保护心肌细胞免受阿霉素诱导的铁死亡
最近的研究表明,铁死亡是一种新型的非凋亡调节性细胞死亡,在阿霉素 (DOX) 诱导的心脏毒性 (DoIC) 中发挥着重要作用。硫化氢(HS)正在成为心血管系统中第三种重要的气体介质。然而,HS 是否对 DOX 诱导的铁死亡有影响仍不清楚。在这里,我们发现在慢性DoIC小鼠模型中,DOX不仅引发心肌细胞铁死亡,而且显着抑制内源性HS的合成。使用HS供体NaHS可明显激活SLC7A11/GSH/GPx4抗氧化途径,从而减轻DOX诱导的小鼠铁死亡和心脏损伤。相比之下,在小鼠 (/Cre) 中,通过心脏特异性敲除胱硫醚 γ-裂解酶基因 () 来消除内源性 HS 的心脏合成,这明显加剧了 DOX 诱导的铁死亡和心脏功能障碍。与具有 DoIC 的 /Cre 小鼠相比,在具有 DoIC 的 /Cre 小鼠中获得了 SLC7A11/GSH/GPx4 通路的进一步抑制。外源性补充 NaHS 可以显着缓解心脏特异性缺乏引起的加重。此外,在 DOX 刺激的 H9c2 心肌细胞中,NaHS 预处理剂量依赖性地增强了 SLC7A11/GSH/GPx4 通路的活性,随后减轻了铁死亡和线粒体损伤。相反,在DOX刺激的H9c2心肌细胞中转染siRNA显着抑制SLC7A11/GSH/GPx4通路,从而导致铁死亡加剧以及线粒体结构和功能的更多损伤。此外,NaHS对DOX诱导的铁死亡的保护作用与S-硫酸化的Keap1密切相关,后者反过来促进Nrf2的核转位以及SLC7A11和GPx4的转录。 总之,我们的研究结果表明,HS 可能通过 Keap1/Nrf2 依赖的 SLC7A11/GSH/GPx4 抗氧化途径抑制 DOX 诱导的铁死亡,从而对 DoIC 发挥保护作用。
更新日期:2024-01-29
中文翻译:
硫化氢通过 Keap1 S-硫化和 Nrf2 激活通过 SLC7A11/GSH/GPx4 途径保护心肌细胞免受阿霉素诱导的铁死亡
最近的研究表明,铁死亡是一种新型的非凋亡调节性细胞死亡,在阿霉素 (DOX) 诱导的心脏毒性 (DoIC) 中发挥着重要作用。硫化氢(HS)正在成为心血管系统中第三种重要的气体介质。然而,HS 是否对 DOX 诱导的铁死亡有影响仍不清楚。在这里,我们发现在慢性DoIC小鼠模型中,DOX不仅引发心肌细胞铁死亡,而且显着抑制内源性HS的合成。使用HS供体NaHS可明显激活SLC7A11/GSH/GPx4抗氧化途径,从而减轻DOX诱导的小鼠铁死亡和心脏损伤。相比之下,在小鼠 (/Cre) 中,通过心脏特异性敲除胱硫醚 γ-裂解酶基因 () 来消除内源性 HS 的心脏合成,这明显加剧了 DOX 诱导的铁死亡和心脏功能障碍。与具有 DoIC 的 /Cre 小鼠相比,在具有 DoIC 的 /Cre 小鼠中获得了 SLC7A11/GSH/GPx4 通路的进一步抑制。外源性补充 NaHS 可以显着缓解心脏特异性缺乏引起的加重。此外,在 DOX 刺激的 H9c2 心肌细胞中,NaHS 预处理剂量依赖性地增强了 SLC7A11/GSH/GPx4 通路的活性,随后减轻了铁死亡和线粒体损伤。相反,在DOX刺激的H9c2心肌细胞中转染siRNA显着抑制SLC7A11/GSH/GPx4通路,从而导致铁死亡加剧以及线粒体结构和功能的更多损伤。此外,NaHS对DOX诱导的铁死亡的保护作用与S-硫酸化的Keap1密切相关,后者反过来促进Nrf2的核转位以及SLC7A11和GPx4的转录。 总之,我们的研究结果表明,HS 可能通过 Keap1/Nrf2 依赖的 SLC7A11/GSH/GPx4 抗氧化途径抑制 DOX 诱导的铁死亡,从而对 DoIC 发挥保护作用。
































 京公网安备 11010802027423号
京公网安备 11010802027423号