当前位置:
X-MOL 学术
›
Nano Today
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Self-assembly of peptides: The acceleration by molecular dynamics simulations and machine learning
Nano Today ( IF 13.2 ) Pub Date : 2024-01-25 , DOI: 10.1016/j.nantod.2024.102160
Nana Cao , Kang Huang , Jianjun Xie , Hui Wang , Xinghua Shi
Nano Today ( IF 13.2 ) Pub Date : 2024-01-25 , DOI: 10.1016/j.nantod.2024.102160
Nana Cao , Kang Huang , Jianjun Xie , Hui Wang , Xinghua Shi
|
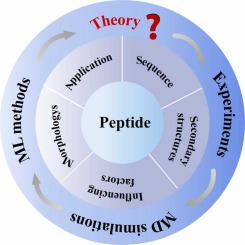
Peptides, biopolymeric compounds connected by peptide bonds, have garnered significant attention in recent years as their potential wide applications in fields such as drug delivery, tissue engineering, and antibiotics. Peptides exhibit excellent biocompatibility and stability due to their structural similarities to many bioactive substances found in human bodies. The self-assembly of peptides has piqued considerable interest with groundbreaking advancements achieved in experimental research. However, it is still a big challenge to establish comprehensive theoretical model to accurately describe the behavior of peptide self-assembly. Current peptide self-assembly designs primarily rely on experimental outcomes and general rules, which is inefficient and susceptible to human errors. In recent years, thanks to rapid advancements in computer techniques and theoretical methods, computational research has become a vital tool in complementing experimental research with rapid development witted in this field. This review delves into the description of peptide self-assembly, covering relevant sequences, structures, morphologies, rules, and application areas. It places particular emphasis on the recent progress in computational methods such as molecular dynamics (MD) simulations and machine learning (ML) techniques in the study. Finally, we provide a perspective on the application of computational methods to expedite exploration in the realm of multi-peptide self-assembly.
中文翻译:

肽的自组装:分子动力学模拟和机器学习的加速
肽是通过肽键连接的生物聚合化合物,近年来因其在药物输送、组织工程和抗生素等领域的潜在广泛应用而受到广泛关注。由于肽与人体中发现的许多生物活性物质的结构相似,因此表现出优异的生物相容性和稳定性。随着实验研究中取得的突破性进展,肽的自组装引起了人们的极大兴趣。然而,建立全面的理论模型来准确描述肽自组装行为仍然是一个巨大的挑战。目前的肽自组装设计主要依赖于实验结果和一般规则,效率低且容易出现人为错误。近年来,由于计算机技术和理论方法的快速进步,计算研究已成为补充实验研究的重要工具,该领域的发展迅速。本综述深入探讨了肽自组装的描述,涵盖相关序列、结构、形态、规则和应用领域。它特别强调研究中分子动力学(MD)模拟和机器学习(ML)技术等计算方法的最新进展。最后,我们提供了应用计算方法来加速多肽自组装领域探索的观点。
更新日期:2024-01-25
中文翻译:
肽的自组装:分子动力学模拟和机器学习的加速
肽是通过肽键连接的生物聚合化合物,近年来因其在药物输送、组织工程和抗生素等领域的潜在广泛应用而受到广泛关注。由于肽与人体中发现的许多生物活性物质的结构相似,因此表现出优异的生物相容性和稳定性。随着实验研究中取得的突破性进展,肽的自组装引起了人们的极大兴趣。然而,建立全面的理论模型来准确描述肽自组装行为仍然是一个巨大的挑战。目前的肽自组装设计主要依赖于实验结果和一般规则,效率低且容易出现人为错误。近年来,由于计算机技术和理论方法的快速进步,计算研究已成为补充实验研究的重要工具,该领域的发展迅速。本综述深入探讨了肽自组装的描述,涵盖相关序列、结构、形态、规则和应用领域。它特别强调研究中分子动力学(MD)模拟和机器学习(ML)技术等计算方法的最新进展。最后,我们提供了应用计算方法来加速多肽自组装领域探索的观点。
































 京公网安备 11010802027423号
京公网安备 11010802027423号