Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adsorption of molecular hydrogen on Be3Al2(SiO3)6-beryl: theoretical insights for catalysis, hydrogen storage, gas separation, sensing, and environmental applications
RSC Advances ( IF 3.9 ) Pub Date : 2024-01-25 , DOI: 10.1039/d3ra07480c Waqas Amber Gill 1 , Norah Alhokbany 2 , Muhammad Ramzan Saeed Ashraf Janjua 3
RSC Advances ( IF 3.9 ) Pub Date : 2024-01-25 , DOI: 10.1039/d3ra07480c Waqas Amber Gill 1 , Norah Alhokbany 2 , Muhammad Ramzan Saeed Ashraf Janjua 3
Affiliation
|
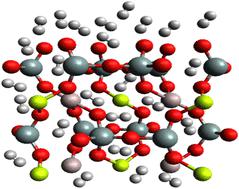
Employing a combination of Density Functional Theory (DFT) calculations and Molecular Dynamics (MD) simulations, the adsorption of molecular hydrogen (H2) on Be3Al2(SiO3)6-beryl, a prominent silicate mineral, has been studied. The crystal structure of beryl, which consists of interconnected tetrahedral and octahedral sites, provides a fascinating framework for comprehending H2 adsorption behavior. Initial investigation of the interaction between H2 molecules and the beryl surface employed DFT calculations. We identified favorable adsorption sites and gained insight into the binding mechanism through extensive structural optimizations and energy calculations. H2 molecules preferentially adsorb on the exposed oxygen atoms surrounding the octahedral sites, producing weak van der Waals interactions with the beryl surface, according to our findings. To further investigate the dynamic aspects of H2 adsorption, MD simulations employing a suitable force field were conducted. To precisely represent interatomic interactions within the Be3Al2(SiO3)6-beryl–H2 system, the force field parameters were meticulously parameterized. By subjecting the system to a variety of temperatures, we were able to obtain valuable information about the stability, diffusion, and desorption kinetics of H2 molecules within the beryl structure. The comprehensive understanding of the H2 adsorption phenomenon on Be3Al2(SiO3)6-beryl is provided by the combined DFT and MD investigations. The results elucidate the mechanisms underlying H2 binding, highlighting the role of surface oxygen atoms and the effect of temperature on H2 dynamics. This research contributes to a fundamental understanding of hydrogen storage and release in beryllium-based silicates and provides valuable guidance for the design and optimization of materials for hydrogen storage, catalysis, gas separation, sensing and environmental applications.
中文翻译:

氢分子吸附在 Be3Al2(SiO3)6-绿柱石上的理论见解:催化、储氢、气体分离、传感和环境应用的理论见解
采用密度泛函理论 (DFT) 计算和分子动力学 (MD) 模拟相结合的方法,研究了氢分子 (H2) 在主要硅酸盐矿物 Be3Al2(SiO3)6-绿柱石上的吸附。绿柱石的晶体结构由相互连接的四面体和八面体位点组成,为理解 H2 吸附行为提供了一个引人入胜的框架。H2 分子与绿柱石表面之间相互作用的初步研究采用了 DFT 计算。我们确定了有利的吸附位点,并通过广泛的结构优化和能量计算深入了解了结合机制。根据我们的研究结果,H2 分子优先吸附在八面体位点周围暴露的氧原子上,与绿柱石表面产生微弱的范德华相互作用。为了进一步研究 H2 吸附的动力学方面,采用合适的力场进行了 MD 模拟。为了精确表示 Be3Al2(SiO3)6-绿柱石-H 2 系统内的原子间相互作用,力场参数被仔细地参数化。通过将系统置于各种温度下,我们能够获得有关绿柱石结构中 H2 分子的稳定性、扩散和解吸动力学的宝贵信息。 DFT 和 MD 联合研究提供了对 Be3Al2(SiO3)6-绿柱石上 H2 吸附现象的全面理解。结果阐明了 H2 结合的潜在机制,突出了表面氧原子的作用和温度对 H2 动力学的影响。这项研究有助于从根本上了解铍基硅酸盐中的氢储存和释放,并为氢储存、催化、气体分离、传感和环境应用的材料设计和优化提供有价值的指导。
更新日期:2024-01-25
中文翻译:
氢分子吸附在 Be3Al2(SiO3)6-绿柱石上的理论见解:催化、储氢、气体分离、传感和环境应用的理论见解
采用密度泛函理论 (DFT) 计算和分子动力学 (MD) 模拟相结合的方法,研究了氢分子 (H2) 在主要硅酸盐矿物 Be3Al2(SiO3)6-绿柱石上的吸附。绿柱石的晶体结构由相互连接的四面体和八面体位点组成,为理解 H2 吸附行为提供了一个引人入胜的框架。H2 分子与绿柱石表面之间相互作用的初步研究采用了 DFT 计算。我们确定了有利的吸附位点,并通过广泛的结构优化和能量计算深入了解了结合机制。根据我们的研究结果,H2 分子优先吸附在八面体位点周围暴露的氧原子上,与绿柱石表面产生微弱的范德华相互作用。为了进一步研究 H2 吸附的动力学方面,采用合适的力场进行了 MD 模拟。为了精确表示 Be3Al2(SiO3)6-绿柱石-H 2 系统内的原子间相互作用,力场参数被仔细地参数化。通过将系统置于各种温度下,我们能够获得有关绿柱石结构中 H2 分子的稳定性、扩散和解吸动力学的宝贵信息。 DFT 和 MD 联合研究提供了对 Be3Al2(SiO3)6-绿柱石上 H2 吸附现象的全面理解。结果阐明了 H2 结合的潜在机制,突出了表面氧原子的作用和温度对 H2 动力学的影响。这项研究有助于从根本上了解铍基硅酸盐中的氢储存和释放,并为氢储存、催化、气体分离、传感和环境应用的材料设计和优化提供有价值的指导。






























 京公网安备 11010802027423号
京公网安备 11010802027423号