当前位置:
X-MOL 学术
›
Eur. J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Synthesis and biological evaluation of selective Pepstatin based trifluoromethylated inhibitors of Cathepsin D
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2024-01-24 , DOI: 10.1016/j.ejmech.2024.116178 Francesco Terzani 1 , Sherazade Belhattab 1 , Aurore Le Guern 1 , Karine Guitot 1 , Olivier Monasson 1 , Chiara Zanato 1 , Evelyne Chelain 1 , Johanne Leroy-Dudal 2 , Julien Pytkowicz 1
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2024-01-24 , DOI: 10.1016/j.ejmech.2024.116178 Francesco Terzani 1 , Sherazade Belhattab 1 , Aurore Le Guern 1 , Karine Guitot 1 , Olivier Monasson 1 , Chiara Zanato 1 , Evelyne Chelain 1 , Johanne Leroy-Dudal 2 , Julien Pytkowicz 1
Affiliation
|
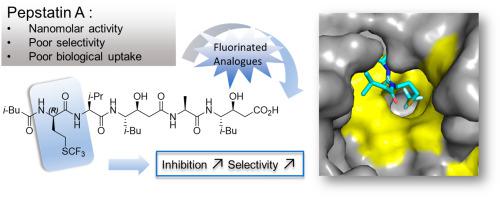
Cathepsin D (CD) is overexpressed in several types of cancer and constitutes an important biological target. Pepstatin A, a pentapeptide incorporating two non-proteinogenic statin residues, is among the most potent inhibitor of CD but lacks selectivity and suffers from poor bioavailability. Eight analogues of Pepstatin A, were synthesized, replacing residues in P3 or P1 position by non-canonical (S)- and (R)-α-Trifluoromethyl Alanine (TfmAla), (S)- and (R)-Trifluoromethionine (TFM) or non-natural -Valine. The biological activities of those analogues were quantified on isolated CD and Pepsin by fluorescence-based assay (FRET) and cytotoxicity of the best fluorinated inhibitors was evaluated on SKOV3 ovarian cancer cell line. (R)-TFM based analog of Pepstatin A (compound ) returned a sub-nanomolar IC50 against CD and an increased selectivity. Molecular Docking experiments could partially rationalize these results. Stabilized inhibitor in the catalytic pocket of CD showed strong hydrophobic interactions of the long and flexible TFM side chain with lipophilic residues of S1 and S3 sub-pockets of the catalytic pocket. The newly synthesized inhibitors returned no cytotoxicity at IC50 concentrations on SKOV3 cancer cells, however the compounds derived from (S)-TfmAla and (R)-TFM led to modifications of cells morphologies, associated with altered organization of F-actin and extracellular Fibronectin.
中文翻译:

选择性胃酶抑素三氟甲基化组织蛋白酶 D 抑制剂的合成及生物学评价
组织蛋白酶 D (CD) 在多种类型的癌症中过度表达,是一个重要的生物学靶点。胃酶抑素 A 是一种含有两个非蛋白原性他汀残基的五肽,是最有效的 CD 抑制剂之一,但缺乏选择性且生物利用度较差。合成了胃酶抑素 A 的八种类似物,用非规范的 (S)- 和 (R)-α-三氟甲基丙氨酸 (TfmAla)、(S)- 和 (R)-三氟甲硫氨酸 (TFM) 替换 P3 或 P1 位上的残基或非天然-缬氨酸。通过基于荧光的测定 (FRET) 对分离的 CD 和胃蛋白酶定量这些类似物的生物活性,并在 SKOV3 卵巢癌细胞系上评估最佳氟化抑制剂的细胞毒性。基于 (R)-TFM 的胃酶抑素 A 类似物(化合物 )返回了针对 CD 的亚纳摩尔 IC50 值和增加的选择性。分子对接实验可以部分合理化这些结果。 CD 催化口袋中的稳定抑制剂显示出长且柔性的 TFM 侧链与催化口袋的 S1 和 S3 子口袋的亲脂残基之间存在强疏水相互作用。新合成的抑制剂在 IC50 浓度下对 SKOV3 癌细胞没有产生细胞毒性,但源自 (S)-TfmAla 和 (R)-TFM 的化合物导致细胞形态发生改变,与 F-肌动蛋白和细胞外纤连蛋白组织的改变有关。
更新日期:2024-01-24
中文翻译:
选择性胃酶抑素三氟甲基化组织蛋白酶 D 抑制剂的合成及生物学评价
组织蛋白酶 D (CD) 在多种类型的癌症中过度表达,是一个重要的生物学靶点。胃酶抑素 A 是一种含有两个非蛋白原性他汀残基的五肽,是最有效的 CD 抑制剂之一,但缺乏选择性且生物利用度较差。合成了胃酶抑素 A 的八种类似物,用非规范的 (S)- 和 (R)-α-三氟甲基丙氨酸 (TfmAla)、(S)- 和 (R)-三氟甲硫氨酸 (TFM) 替换 P3 或 P1 位上的残基或非天然-缬氨酸。通过基于荧光的测定 (FRET) 对分离的 CD 和胃蛋白酶定量这些类似物的生物活性,并在 SKOV3 卵巢癌细胞系上评估最佳氟化抑制剂的细胞毒性。基于 (R)-TFM 的胃酶抑素 A 类似物(化合物 )返回了针对 CD 的亚纳摩尔 IC50 值和增加的选择性。分子对接实验可以部分合理化这些结果。 CD 催化口袋中的稳定抑制剂显示出长且柔性的 TFM 侧链与催化口袋的 S1 和 S3 子口袋的亲脂残基之间存在强疏水相互作用。新合成的抑制剂在 IC50 浓度下对 SKOV3 癌细胞没有产生细胞毒性,但源自 (S)-TfmAla 和 (R)-TFM 的化合物导致细胞形态发生改变,与 F-肌动蛋白和细胞外纤连蛋白组织的改变有关。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号