当前位置:
X-MOL 学术
›
Adv. Energy Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Asymmetric Copper-Sulphur Sites Promote C–C Coupling for Selective CO2 Electroreduction to C2 Products
Advanced Energy Materials ( IF 24.4 ) Pub Date : 2024-01-21 , DOI: 10.1002/aenm.202304224
Liang Liang 1 , Li Yang 2, 3 , Thomas Heine 2, 3 , Aleks Arinchtein 1 , Xingli Wang 1 , Jessica Hübner 1 , Johannes Schmidt 4 , Arne Thomas 4 , Peter Strasser 1
Advanced Energy Materials ( IF 24.4 ) Pub Date : 2024-01-21 , DOI: 10.1002/aenm.202304224
Liang Liang 1 , Li Yang 2, 3 , Thomas Heine 2, 3 , Aleks Arinchtein 1 , Xingli Wang 1 , Jessica Hübner 1 , Johannes Schmidt 4 , Arne Thomas 4 , Peter Strasser 1
Affiliation
|
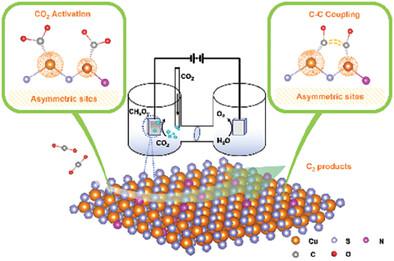
Sustainable multicarbon e-chemicals are of particular interest due to their potential future, high market values, and demand. In the direct electrocatalytic formation of multicarbon e-chemicals from CO2, the elementary C–C coupling by CO dimerization is considered the rate-limiting step. Here, a generalized surface structural design principle of asymmetric metal pair sites is proposed, explored, and experimentally tested in order to promote CO dimerization on surfaces. First a computational model of N-doped Cu2S layers featuring adjacent, electronically asymmetric Cuδ1+-Cuδ2+ (0 < δ1+ < δ2+ < 1) metal atomic pairs evidenced by their non-uniform charge distribution is considered. The electronic asymmetry resulted in distinct CO adsorption energies and the associated self-adjusting structures, which lowered C–C coupling energy barriers significantly. The computational hypotheses are experimentally tested using X-ray photoelectron spectroscopy of Cu-N moieties within N-doped Cu2S layers. In-situ Fourier-transform infrared spectroscopy confirms linear *CO and *CO-CO adsorption configuration by the peaks of ≈2080 and 1920 cm−1, respectively. After N-doping, the catalytically C2 faradaic efficiency can significantly be elevated to 14.72% due to the promotion of C–C coupling.
中文翻译:

不对称铜硫位点促进 C-C 耦合,选择性 CO2 电还原为 C2 产品
可持续多碳电子化学品因其潜在的未来、高市场价值和需求而受到特别关注。在从 CO 2直接电催化形成多碳电子化学品的过程中,CO 二聚的基本 C-C 偶联被认为是限速步骤。在这里,提出、探索和实验测试了不对称金属对位点的广义表面结构设计原理,以促进表面上的CO二聚化。首先考虑 N 掺杂 Cu 2 S 层的计算模型,该模型具有相邻的、电子不对称的 Cu δ1+ -Cu δ2+ (0 < δ 1 + < δ 2 + < 1) 金属原子对,这由它们的非均匀电荷分布证明。电子不对称性导致不同的CO吸附能和相关的自调节结构,从而显着降低了C-C耦合能垒。使用N掺杂Cu 2 S层内的Cu-N部分的X射线光电子能谱对计算假设进行了实验测试。原位傅里叶变换红外光谱分别通过约2080和1920 cm -1的峰确认了线性*CO和*CO-CO吸附构型。 N掺杂后,由于C-C耦合的促进,催化C 2法拉第效率可显着提高至14.72%。
更新日期:2024-01-21
中文翻译:
不对称铜硫位点促进 C-C 耦合,选择性 CO2 电还原为 C2 产品
可持续多碳电子化学品因其潜在的未来、高市场价值和需求而受到特别关注。在从 CO 2直接电催化形成多碳电子化学品的过程中,CO 二聚的基本 C-C 偶联被认为是限速步骤。在这里,提出、探索和实验测试了不对称金属对位点的广义表面结构设计原理,以促进表面上的CO二聚化。首先考虑 N 掺杂 Cu 2 S 层的计算模型,该模型具有相邻的、电子不对称的 Cu δ1+ -Cu δ2+ (0 < δ 1 + < δ 2 + < 1) 金属原子对,这由它们的非均匀电荷分布证明。电子不对称性导致不同的CO吸附能和相关的自调节结构,从而显着降低了C-C耦合能垒。使用N掺杂Cu 2 S层内的Cu-N部分的X射线光电子能谱对计算假设进行了实验测试。原位傅里叶变换红外光谱分别通过约2080和1920 cm -1的峰确认了线性*CO和*CO-CO吸附构型。 N掺杂后,由于C-C耦合的促进,催化C 2法拉第效率可显着提高至14.72%。


































 京公网安备 11010802027423号
京公网安备 11010802027423号