Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2024-01-18 , DOI: 10.1016/j.molstruc.2024.137552
Melisa Kiran , Zeynep Pinar Haslak , Halit Ates , Viktorya Aviyente , Fatma Ahu Akin
|
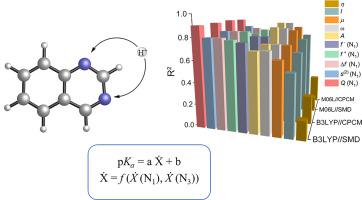
In this study, several quantum mechanical-based computational approaches have been used in order to propose accurate protocols for predicting the pKa’s of quinazoline derivatives, which constitute a very important class of natural and synthetic compounds in organic, pharmaceutical, agricultural and medicinal chemistry areas. Linear relationships between the experimental pKa’s and nine different DFT descriptors (atomic charge on nitrogen atoms (Q(N)), ionization energy (I), electron affinity (A), chemical potential (μ), hardness (η), electrophilicity index (ω), fukui functions (f +, f −), condensed dual descriptor (Δf) and local hypersoftness () were considered. Several DFT methods (a combination of five DFT functionals and two basis sets) in conjunction with two different implicit solvent models were tested, and among them, M06L/6–311++G(d,p) level of theory employing the CPCM solvation model was found to give the strongest correlations between the DFT descriptors and the experimental pKa’s of the quinazoline derivatives. The calculated atomic charge on N1 atom (Q(N1)) was shown to be the best descriptor to reproduce the experimental pKa’s (R2=0.927), whereas strong correlations were also derived for A, ω, μ, and Δf. The QM-based protocols presented in this study will enable fast and accurate high-throughput pKa predictions of quinazoline derivatives and the relationships derived can be effectively used in data generation for successful machine learning models for pKa predictions.
中文翻译:
喹唑啉衍生物的 pKa 评估:基于量子力学的描述符的使用
在这项研究中,使用了几种基于量子力学的计算方法,以提出准确的方案来预测喹唑啉衍生物的 p K a,喹唑啉衍生物构成有机、制药、农业和农业领域中一类非常重要的天然和合成化合物。药物化学领域。实验 p K a与九种不同 DFT 描述符(氮原子上的原子电荷 ( Q (N))、电离能 ( I )、电子亲和力 ( A )、化学势 (μ)、硬度 (η)之间的线性关系、亲电指数 (ω)、福井函数 ( f + , f − )、压缩对偶描述符 (Δ f ) 和局部超柔软度 () 被认为是。测试了几种 DFT 方法(五个 DFT 泛函和两个基组的组合)与两种不同隐式溶剂模型的结合,其中 M06L/6–311++ G (d,p) 级别的理论采用 CPCM 溶剂化发现该模型在 DFT 描述符和喹唑啉衍生物的实验pKa之间具有最强的相关性。计算出的 N 1原子上的原子电荷 ( Q (N 1 )) 被证明是重现实验 p Ka 的最佳描述符( R 2 = 0.927),而A、 ω 、 μ也得出了强相关性,和Δf。本研究中提出的基于 QM 的协议将能够快速、准确地对喹唑啉衍生物进行高通量 p K a预测,并且所得出的关系可以有效地用于数据生成,以实现 p K a预测的成功机器学习模型。


































 京公网安备 11010802027423号
京公网安备 11010802027423号