Scientific Reports ( IF 3.8 ) Pub Date : 2024-01-18 , DOI: 10.1038/s41598-024-51958-z Ali Karimnezhad 1, 2 , Theodore J Perkins 3, 4
|
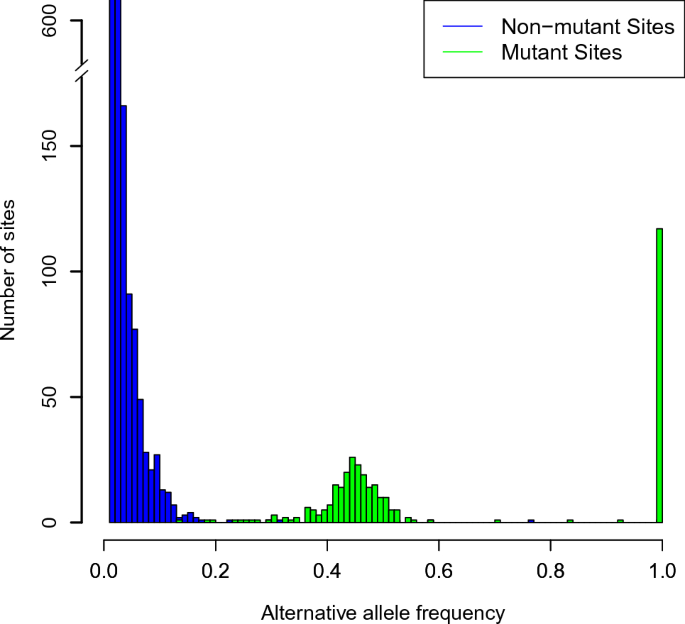
One of the fundamental computational problems in cancer genomics is the identification of single nucleotide variants (SNVs) from DNA sequencing data. Many statistical models and software implementations for SNV calling have been developed in the literature, yet, they still disagree widely on real datasets. Based on an empirical Bayesian approach, we introduce a local false discovery rate (LFDR) estimator for germline SNV calling. Our approach learns model parameters without prior information, and simultaneously accounts for information across all sites in the genomic regions of interest. We also propose another LFDR-based algorithm that reliably prioritizes a given list of mutations called by any other variant-calling algorithm. We use a suite of gold-standard cell line data to compare our LFDR approach against a collection of widely used, state of the art programs. We find that our LFDR approach approximately matches or exceeds the performance of all of these programs, despite some very large differences among them. Furthermore, when prioritizing other algorithms’ calls by our LFDR score, we find that by manipulating the type I-type II tradeoff we can select subsets of variant calls with minimal loss of sensitivity but dramatic increases in precision.
中文翻译:
下一代测序数据的经验贝叶斯单核苷酸变异调用
癌症基因组学中的基本计算问题之一是从 DNA 测序数据中识别单核苷酸变异 (SNV)。文献中已经开发了许多用于 SNV 调用的统计模型和软件实现,但它们在实际数据集上仍然存在广泛分歧。基于经验贝叶斯方法,我们引入了一种用于种系 SNV 调用的局部错误发现率 (LFDR) 估计器。我们的方法无需先验信息即可学习模型参数,并同时考虑感兴趣的基因组区域中所有位点的信息。我们还提出了另一种基于 LFDR 的算法,该算法可靠地对由任何其他变体调用算法调用的给定突变列表进行优先级排序。我们使用一套黄金标准细胞系数据将我们的 LFDR 方法与一系列广泛使用的最先进的程序进行比较。我们发现我们的 LFDR 方法大致匹配或超过所有这些程序的性能,尽管它们之间存在很大的差异。此外,当通过 LFDR 分数对其他算法的调用进行优先级排序时,我们发现通过操纵 I 型-II 型权衡,我们可以选择变体调用的子集,同时灵敏度损失最小,但精度显着提高。






























 京公网安备 11010802027423号
京公网安备 11010802027423号