当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Which Electronic Structure Method to Choose in Trajectory Surface Hopping Dynamics Simulations? Azomethane as a Case Study
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2024-01-11 , DOI: 10.1021/acs.jpclett.3c03014
Thomas V Papineau 1 , Denis Jacquemin 1, 2 , Morgane Vacher 1
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2024-01-11 , DOI: 10.1021/acs.jpclett.3c03014
Thomas V Papineau 1 , Denis Jacquemin 1, 2 , Morgane Vacher 1
Affiliation
|
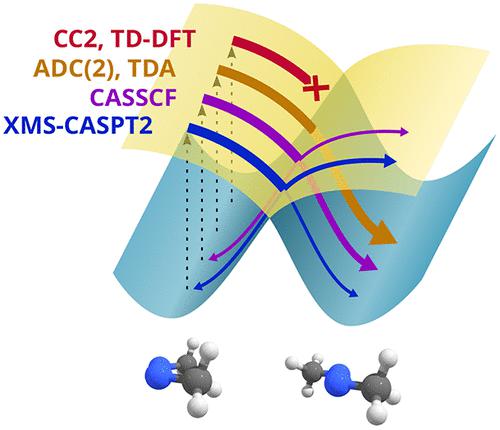
Nonadiabatic dynamics simulations have become a standard approach to explore photochemical reactions. Such simulations require underlying potential energy surfaces and couplings between them, calculated at a chosen level of theory, yet this aspect is rarely assessed. Here, in combination with the popular trajectory surface hopping dynamics method, we use a high-accuracy XMS-CASPT2 electronic structure level as a benchmark for assessing the performances of various post-Hartree–Fock methods (namely, CIS, ADC(2), CC2, and CASSCF) and exchange–correlation functionals (PBE, PBE0, and CAM-B3LYP) in a TD-DFT/TDA context, using the isomerization around a double bond as test case. Different relaxation pathways are identified, and the ability of the different methods to reproduce their relative importance and time scale is discussed. The results show that multireference electronic structure methods should be preferred, when studying nonadiabatic decay between excited and ground states. If not affordable, TD-DFT with TDA and hybrid functionals and ADC(2) are efficient alternatives but overestimate the nonradiative decay yield and thus may miss deexcitation pathways.
中文翻译:

在弹道表面跳跃动力学模拟中选择哪种电子结构方法?偶氮甲烷作为案例研究
非绝热动力学模拟已成为探索光化学反应的标准方法。这种模拟需要潜在的势能表面以及它们之间的耦合,在选定的理论水平上进行计算,但这方面很少被评估。在这里,结合流行的轨迹表面跳跃动力学方法,我们使用高精度的XMS-CASPT2电子结构水平作为评估各种后Hartree-Fock方法(即CIS、ADC(2)、 CC2 和 CASSCF)和 TD-DFT/TDA 环境中的交换相关泛函(PBE、PBE0 和 CAM-B3LYP),使用围绕双键的异构化作为测试用例。确定了不同的松弛途径,并讨论了不同方法重现其相对重要性和时间尺度的能力。结果表明,在研究激发态和基态之间的非绝热衰变时,应优先考虑多参考电子结构方法。如果负担不起,具有 TDA 和混合泛函的 TD-DFT 以及 ADC(2) 是有效的替代方案,但会高估非辐射衰减率,因此可能会错过去激励路径。
更新日期:2024-01-11
中文翻译:
在弹道表面跳跃动力学模拟中选择哪种电子结构方法?偶氮甲烷作为案例研究
非绝热动力学模拟已成为探索光化学反应的标准方法。这种模拟需要潜在的势能表面以及它们之间的耦合,在选定的理论水平上进行计算,但这方面很少被评估。在这里,结合流行的轨迹表面跳跃动力学方法,我们使用高精度的XMS-CASPT2电子结构水平作为评估各种后Hartree-Fock方法(即CIS、ADC(2)、 CC2 和 CASSCF)和 TD-DFT/TDA 环境中的交换相关泛函(PBE、PBE0 和 CAM-B3LYP),使用围绕双键的异构化作为测试用例。确定了不同的松弛途径,并讨论了不同方法重现其相对重要性和时间尺度的能力。结果表明,在研究激发态和基态之间的非绝热衰变时,应优先考虑多参考电子结构方法。如果负担不起,具有 TDA 和混合泛函的 TD-DFT 以及 ADC(2) 是有效的替代方案,但会高估非辐射衰减率,因此可能会错过去激励路径。






























 京公网安备 11010802027423号
京公网安备 11010802027423号