当前位置:
X-MOL 学术
›
J. Mol. Liq.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Solvent effects on the structural, spectroscopic, electronic properties, NCI-RDG analysis, molecular docking and molecular dynamics studies of 1-benzyl-indole-3-carbinol
Journal of Molecular Liquids ( IF 5.3 ) Pub Date : 2024-01-07 , DOI: 10.1016/j.molliq.2024.123967 Stève-Jonathan Koyambo-Konzapa , R. Premkumar , George Amolo , Mama Nsangou
Journal of Molecular Liquids ( IF 5.3 ) Pub Date : 2024-01-07 , DOI: 10.1016/j.molliq.2024.123967 Stève-Jonathan Koyambo-Konzapa , R. Premkumar , George Amolo , Mama Nsangou
|
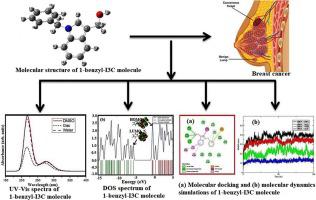
This investigation focuses on the 1-benzyl-indole-3-carbinol (1-benzyl-I3C) molecule and its capacity as a drug to combat breast cancer. The research employs different computational approaches including DFT simulations, molecular docking, and molecular dynamics calculations. The molecular configuration of 1-benzyl-I3C was optimized in the COSMO solvation model, using B3LYP/cc-pVTZ basis set. The modes of vibration of the compound were identified through Potential Energy Distribution (PED) simulations, and the results were found to be consistent with the available FT-IR and Raman data. The analysis of Natural Bond Orbitals (NBO) supports the idea that 1-benzyl-I3C has bioactive properties. The impact of solvents on the electronic characteristics of studied molecule is discussed. The reactive sites of 1-benzyl-I3C were confirmed by the local reactivity descriptors and the molecular electrostatic potential (MEP) surface analyses. Additionally, Non-covalent interaction (NCI) analysis confirms the existence of the Van der Waals interactions in the compound under investigation. Anti-breast cancer activities of the 1-benzyl-I3C molecule were examined based on molecular docking analysis, which reveals a good inhibitor nature of the studied molecule against the progesterone receptor (PR). The molecular dynamics analysis through the RMSD, RMSF, and Rg plots confirms the overall stability of the obtained 1-benzyl-I3C-PR complex.
中文翻译:

溶剂对 1-苄基吲哚-3-甲醇的结构、光谱、电子性质、NCI-RDG 分析、分子对接和分子动力学研究的影响
这项研究的重点是 1-benzyl-indole-3-carbinol (1-benzyl-I3C) 分子及其作为抗乳腺癌药物的能力。该研究采用了不同的计算方法,包括 DFT 模拟、分子对接和分子动力学计算。使用 B3LYP/cc-pVTZ 基本组在 COSMO 溶剂化模型中优化 1-benzyl-I3C 的分子构型。通过势能分布 (PED) 模拟确定了化合物的振动模式,结果与现有的 FT-IR 和拉曼数据一致。自然键轨道 (NBO) 的分析支持了 1-benzyl-I3C 具有生物活性的观点。讨论了溶剂对所研究分子的电子特性的影响。通过局部反应描述符和分子静电势 (MEP) 表面分析证实了 1-benzyl-I3C 的反应位点。此外,非共价相互作用 (NCI) 分析证实了所研究的化合物中存在范德华相互作用。基于分子对接分析检查了 1-benzyl-I3C 分子的抗乳腺癌活性,揭示了所研究的分子对孕酮受体(PR)具有良好的抑制剂性质。通过 RMSD、RMSF 和 Rg 图进行的分子动力学分析证实了所获得的 1-苄基-I3C-PR 复合物的整体稳定性。
更新日期:2024-01-07
中文翻译:
溶剂对 1-苄基吲哚-3-甲醇的结构、光谱、电子性质、NCI-RDG 分析、分子对接和分子动力学研究的影响
这项研究的重点是 1-benzyl-indole-3-carbinol (1-benzyl-I3C) 分子及其作为抗乳腺癌药物的能力。该研究采用了不同的计算方法,包括 DFT 模拟、分子对接和分子动力学计算。使用 B3LYP/cc-pVTZ 基本组在 COSMO 溶剂化模型中优化 1-benzyl-I3C 的分子构型。通过势能分布 (PED) 模拟确定了化合物的振动模式,结果与现有的 FT-IR 和拉曼数据一致。自然键轨道 (NBO) 的分析支持了 1-benzyl-I3C 具有生物活性的观点。讨论了溶剂对所研究分子的电子特性的影响。通过局部反应描述符和分子静电势 (MEP) 表面分析证实了 1-benzyl-I3C 的反应位点。此外,非共价相互作用 (NCI) 分析证实了所研究的化合物中存在范德华相互作用。基于分子对接分析检查了 1-benzyl-I3C 分子的抗乳腺癌活性,揭示了所研究的分子对孕酮受体(PR)具有良好的抑制剂性质。通过 RMSD、RMSF 和 Rg 图进行的分子动力学分析证实了所获得的 1-苄基-I3C-PR 复合物的整体稳定性。







































 京公网安备 11010802027423号
京公网安备 11010802027423号