Interdisciplinary Sciences: Computational Life Sciences ( IF 3.9 ) Pub Date : 2024-01-08 , DOI: 10.1007/s12539-023-00597-5 Minghua Hou 1 , Sirong Jin 1 , Xinyue Cui 1 , Chunxiang Peng 1 , Kailong Zhao 1 , Le Song 2 , Guijun Zhang 1
|
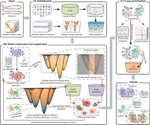
The breakthrough of AlphaFold2 and the publication of AlphaFold DB represent a significant advance in the field of predicting static protein structures. However, AlphaFold2 models tend to represent a single static structure, and multiple-conformation prediction remains a challenge. In this work, we proposed a method named MultiSFold, which uses a distance-based multi-objective evolutionary algorithm to predict multiple conformations. To begin, multiple energy landscapes are constructed using different competing constraints generated by deep learning. Subsequently, an iterative modal exploration and exploitation strategy is designed to sample conformations, incorporating multi-objective optimization, geometric optimization and structural similarity clustering. Finally, the final population is generated using a loop-specific sampling strategy to adjust the spatial orientations. MultiSFold was evaluated against state-of-the-art methods using a benchmark set containing 80 protein targets, each characterized by two representative conformational states. Based on the proposed metric, MultiSFold achieves a remarkable success ratio of 56.25% in predicting multiple conformations, while AlphaFold2 only achieves 10.00%, which may indicate that conformational sampling combined with knowledge gained through deep learning has the potential to generate conformations spanning the range between different conformational states. In addition, MultiSFold was tested on 244 human proteins with low structural accuracy in AlphaFold DB to test whether it could further improve the accuracy of static structures. The experimental results demonstrate the performance of MultiSFold, with a TM-score better than that of AlphaFold2 by 2.97% and RoseTTAFold by 7.72%. The online server is at http://zhanglab-bioinf.com/MultiSFold.
Graphical Abstract
中文翻译:
使用多目标进化算法预测蛋白质多构象
AlphaFold2的突破和AlphaFold DB的发布代表了预测静态蛋白质结构领域的重大进展。然而,AlphaFold2 模型往往表示单一静态结构,多构象预测仍然是一个挑战。在这项工作中,我们提出了一种名为 MultiSFold 的方法,该方法使用基于距离的多目标进化算法来预测多种构象。首先,使用深度学习产生的不同竞争约束构建多个能源景观。随后,设计了迭代模态探索和开发策略来采样构象,结合多目标优化、几何优化和结构相似性聚类。最后,使用特定于循环的采样策略来调整空间方向来生成最终总体。使用包含 80 个蛋白质靶标的基准集,根据最先进的方法对 MultiSFold 进行评估,每个靶标都有两种代表性的构象状态。基于所提出的指标,MultiSFold 在预测多种构象方面取得了 56.25% 的显着成功率,而 AlphaFold2 仅达到 10.00%,这可能表明构象采样与通过深度学习获得的知识相结合有潜力生成跨越范围的构象不同的构象状态。此外,MultiSFold还在AlphaFold DB中对244种结构精度较低的人类蛋白质进行了测试,以测试其是否可以进一步提高静态结构的精度。实验结果证明了MultiSFold的性能,其TM分数比AlphaFold2好2.97%,比RoseTTAFold好7.72%。 在线服务器位于http://zhanglab-bioinf.com/MultiSFold。






























 京公网安备 11010802027423号
京公网安备 11010802027423号