Journal of Molecular Modeling ( IF 2.1 ) Pub Date : 2024-01-02 , DOI: 10.1007/s00894-023-05812-0 Jeanet Conradie 1, 2
|
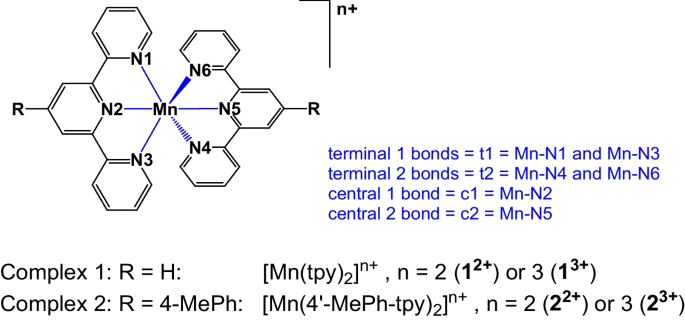
Context
Bis(terpyridine)manganese(III) exhibits Jahn–Teller distortion due to the inequivalent occupation of the degenerate eg orbitals of this high-spin d4 pseudo octahedral complex. Due to the spatially constrained nature of the terpyridine ligand, the central Mn-N bonds will always be shorter than the Mn-N terminal bonds, making it more difficult to distinguish between compression and elongation Jahn–Teller structures for bis(terpyridine)manganese(III). Density functional theory (DFT) calculations were utilized as a tool to evaluate the type of Jahn–Teller distortion in the high-spin d4 bis(terpyridine)manganese(III). The nature of the Jahn–Teller distortion calculated does depend upon the choice of density functional approximation (DFA) with the B3LYP, M06, and OLYP-D3 DFAs giving compression and the PW6B95D3, MN15, and MN15-D3 DFAs giving elongation in gas-phase calculations. All solvent-phase calculations yield an elongated structure for the bis(terpyridine)manganese(III) compound, which is yet to be structurally characterized experimentally. However, both gas and solvent OLYP-D3 calculations result in a compressed structure for the only experimentally isolated and characterized bis(terpyridine)manganese(III) complex, specifically the complex with terpyridine = 4′-(4-methylphenyl)-2,2′:6′,2′′-terpyridine. This alignment with the experimentally observed compression Jahn–Teller structure enhances the credibility of OLYP-D3 calculations in reproducing the observed geometries. The compressed Jahn–Teller geometries were near D2d symmetry with the z-axis for compression defined along the Mn-N central bonds. Elongation Jahn–Teller distortion is not possible along the Mn-N central bonds, due to their spatially constrained nature. Thus, elongation occur along one pair of opposite Mn-N terminal bonds that are longer than the other pair of opposite terminal bonds, with shorter central bonds. The highest symmetry of the elongation Jahn–Teller distortion geometry of bis(terpyridine)manganese(III) is C2v. Criteria to distinguish between a compression and elongation Jahn–Teller geometry for bis(terpyridine)manganese(III) are identified. The nature of the singly occupied eg molecular orbital, exhibiting anti-bonding interaction with the nitrogen-p MOs involved, dictates the type of Jahn–Teller distortion that occurs. The low-energy occupied bonding t2g molecular orbitals establish bonds with and undergo mixing with the ligand molecular orbitals. The OLYP-D3 functional is recommended for calculating bis(terpyridine)manganese(III) and related compounds due to its consistent generation of metal–ligand bonds slightly longer than observed in experiments, in line with the required behavior. Additionally, OLYP-D3 offers a realistic electronic structure for Jahn–Teller distorted bis(terpyridine)manganese(III), correctly identifying alpha eg molecular orbitals as the highest occupied molecular orbital and lowest unoccupied molecular orbital in agreement with experimental electrochemical studies. Furthermore, OLYP-D3 accurately reproduces the experimental compression geometry for the only structurally known bis(terpyridine)manganese(III) compound, instilling confidence in its reliability for such calculations.
Methods
DFT geometry optimization and frequency calculations were done on the two different modes of Jahn–Teller distortion of bis(terpyridine)manganese(III), using the OLYP, B3LYP, M06, PW6B95D3, and MN15 functionals, with and without the Grimme’s D3 dispersion correction, and the 6-311G(d,p) or def2TZVPP basis set, as implemented in Gaussian 16. All optimizations were in the gas phase and also in the solvent phase with CH3CN as implicit solvent using IEFPCM.
Graphical Abstract
DFT calculations were utilized to determine the Jahn–Teller effect on the geometry of high-spin d4 bis(terpyridine)manganese(III) complex containing two structurally constrained tridentate ligands.
中文翻译:
密度泛函近似对双(三联吡啶)锰(III)及相关化合物计算的Jahn-Teller畸变的影响
语境
双(三联吡啶)锰(III)由于这种高自旋d 4伪八面体络合物的简并e g轨道的不等占据而表现出Jahn-Teller畸变。由于三联吡啶配体的空间受限性质,中心 Mn-N 键总是比 Mn-N 末端键短,这使得区分双(三联吡啶)锰的压缩和伸长 Jahn-Teller 结构变得更加困难。三)。密度泛函理论 (DFT) 计算被用作评估高自旋 d 4双(三联吡啶)锰 (III) 中 Jahn-Teller 畸变类型的工具。计算出的 Jahn-Teller 畸变的性质确实取决于密度泛函近似 (DFA) 的选择,其中 B3LYP、M06 和 OLYP-D3 DFA 提供压缩,PW6B95D3、MN15 和 MN15-D3 DFA 提供气体伸长。相位计算。所有溶剂相计算都会产生双(三联吡啶)锰(III)化合物的细长结构,该结构尚未通过实验进行结构表征。然而,气体和溶剂 OLYP-D3 计算均得出了唯一经过实验分离和表征的双(三联吡啶)锰(III)络合物的压缩结构,特别是具有三联吡啶 = 4'-(4-甲基苯基)-2,2 的络合物':6',2''-三联吡啶。这种与实验观察到的压缩 Jahn-Teller 结构的一致性增强了 OLYP-D3 计算在再现观察到的几何形状方面的可信度。压缩的 Jahn-Teller 几何形状接近D 2d对称性,压缩z轴沿 Mn-N 中心键定义。 由于 Mn-N 中心键的空间受限性质,伸长 Jahn-Teller 畸变是不可能发生的。因此,伸长沿着一对相对的 Mn-N 末端键发生,该对 Mn-N 末端键比另一对相对的末端键长,且中心键较短。双(三联吡啶)锰(III)伸长Jahn-Teller畸变几何的最高对称性是C 2v 。确定了区分双(三联吡啶)锰(III)压缩和伸长 Jahn-Teller 几何形状的标准。单占据例如分子轨道的性质,表现出与所涉及的氮-p MOs的反键相互作用,决定了发生的Jahn-Teller畸变的类型。低能占据键合t 2g分子轨道与配体分子轨道建立键合并进行混合。建议使用 OLYP-D3 泛函计算双(三联吡啶)锰(III)和相关化合物,因为它一致生成的金属-配体键略长于实验中观察到的长度,符合所需的行为。此外,OLYP-D3 为 Jahn-Teller 扭曲双(三联吡啶)锰(III)提供了真实的电子结构,正确地将 α e g分子轨道识别为最高占据分子轨道和最低未占据分子轨道,与实验电化学研究一致。此外,OLYP-D3 准确地再现了唯一结构已知的双(三联吡啶)锰(III)化合物的实验压缩几何形状,使其对此类计算的可靠性充满信心。
方法
使用 OLYP、B3LYP、M06、PW6B95D3 和 MN15 泛函,在有和没有 Grimme's D3 色散校正的情况下,对双(三联吡啶)锰(III)的 Jahn-Teller 畸变的两种不同模式进行了 DFT 几何优化和频率计算,以及 6-311G(d,p) 或 def2TZVPP 基组,如 Gaussian 16 中所实现。所有优化均在气相中进行,也在溶剂相中进行,使用 IEFPCM 将 CH 3 CN 作为隐式溶剂。
图形概要
DFT 计算用于确定 Jahn-Teller 对包含两个结构受限的三齿配体的高自旋 d 4双(三联吡啶)锰(III)配合物几何形状的影响。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号