Scientific Reports ( IF 3.8 ) Pub Date : 2023-12-27 , DOI: 10.1038/s41598-023-50427-3 Hiroaki Tatsumi, C. R. Kao, Hiroshi Nishikawa
|
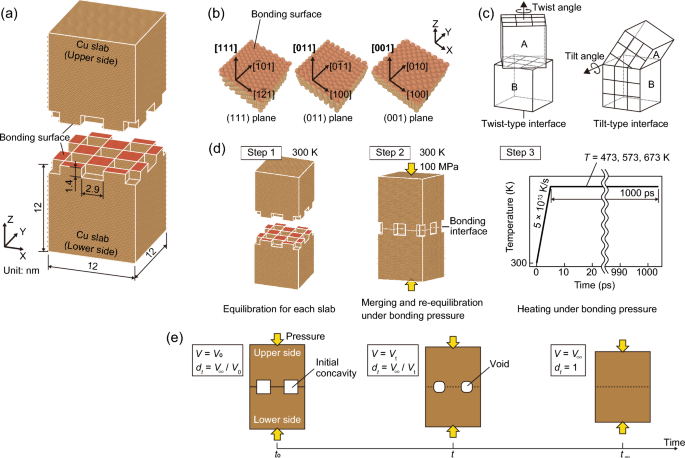
High-density electronics are hindered by the constraints of Sn-based solder joints, necessitating the exploration of Cu–Cu solid-state bonding. However, current bonding methods are expensive and time-consuming; therefore, understanding the Cu–Cu bonding mechanism is crucial for optimization. This study utilizes molecular dynamics (MD) simulation to elucidate the Cu–Cu solid-state bonding behavior, focusing on interfacial densification and diffusion phenomena. Furthermore, it highlights the influence of crystal orientation on the interfacial bonding behavior. To analyze the impact of crystal orientation, monocrystalline Cu slabs with a simplified periodic surface structure were employed to replicate surface roughness and subsequently bonded at a specific temperature. The results indicate the critical influence of crystalline orientations on the bonding process: identical orientations result in slower densification at the interface, whereas misoriented orientations significantly accelerate it. This effect, attributed to the grain boundary (GB) structures formed owing to misorientation, suggests a central role for GB diffusion in bonding progression. Diffusion coefficients calculated using the mean square displacement (MSD) confirmed these findings and exhibited significantly larger values for misoriented joints. Additionally, the simulations reveal an activation energy for GB diffusion that is lower than conventional values, highlighting the impact of the crystallographic orientation and voids at the bonding interface. Our research elucidates the role of crystalline orientation in diffusion phenomena at bonding interfaces, offering valuable implications for optimizing bonding-based manufacturing processes.
中文翻译:
通过分子动力学模拟晶体取向对 Cu-Cu 固态键合行为的影响
高密度电子器件受到锡基焊点的限制,因此需要探索铜-铜固态键合。然而,目前的粘合方法既昂贵又耗时;因此,了解 Cu-Cu 键合机制对于优化至关重要。本研究利用分子动力学 (MD) 模拟来阐明 Cu-Cu 固态键合行为,重点关注界面致密化和扩散现象。此外,它强调了晶体取向对界面键合行为的影响。为了分析晶体取向的影响,采用具有简化周期性表面结构的单晶铜板来复制表面粗糙度,然后在特定温度下进行粘合。结果表明晶体取向对键合过程的关键影响:相同的取向导致界面致密化速度减慢,而错误取向的取向则显着加速其致密化。这种效应归因于由于取向错误而形成的晶界(GB)结构,表明晶界扩散在键合进展中发挥着核心作用。使用均方位移(MSD)计算的扩散系数证实了这些发现,并且对于定向错误的关节表现出明显更大的值。此外,模拟显示晶界扩散的活化能低于传统值,突出了键合界面处的晶体取向和空隙的影响。我们的研究阐明了晶体取向在键合界面扩散现象中的作用,为优化基于键合的制造工艺提供了宝贵的启示。





























 京公网安备 11010802027423号
京公网安备 11010802027423号