Molecular Catalysis ( IF 3.9 ) Pub Date : 2023-12-24 , DOI: 10.1016/j.mcat.2023.113795 Rong Fan , Ruiying Li , Xinping Wang , Xinhua Kuang , Jiabao Li , Yibin Liu , Hao Yan , Xin Zhou , Hui Zhao , Xiang Feng , Xiaobo Chen , Chaohe Yang
|
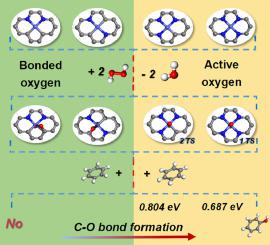
Adjusting the local environment of single-atom catalysts (SACs) is a promising approach to enhance the catalytic performance in benzene oxidation reaction (BOR). Herein, a range of Co-based SACs with N2/3/4-coordination numbers on graphene were constructed to investigate the effect of the electronic structure of SACs on BOR via density function theory (DFT). Expectedly, the electron density and charge amount of the Co active sites in the overall reaction path on the CoNxCy catalyst can be regulated by coordination environment. In particular, the as-formed *O=CoN4=O* intermediate displays the strongest electron interaction between Co atom and O*, thus O* has the largest electron density and the most negative charge number, resulting in a lower energy barrier of 0.69 eV for the rate-determining step of C O bond formation. The methodology in this work provides a certain theory supporting and guidance for developing highly efficient SACs with controllable coordination structures to enhance hydrocarbon oxidation reactions.
O bond formation. The methodology in this work provides a certain theory supporting and guidance for developing highly efficient SACs with controllable coordination structures to enhance hydrocarbon oxidation reactions.
中文翻译:
Co-NxCy结构苯选择性氧化制苯酚局部环境的理论研究
调整单原子催化剂(SAC)的局部环境是提高苯氧化反应(BOR)催化性能的一种有前景的方法。在此,构建了一系列在石墨烯上具有N 2/3/4配位数的Co基SAC,以通过密度函数理论(DFT)研究SAC的电子结构对BOR的影响。预计CoN x C y催化剂上整个反应路径中Co活性位点的电子密度和电荷量可以通过配位环境来调节。特别是,所形成的 *O=CoN 4 =O* 中间体显示出 Co 原子与 O* 之间最强的电子相互作用,因此 O* 具有最大的电子密度和最多的负电荷数,从而导致较低的能垒0.69 eV 用于 CO 键形成的速率决定步骤。这项工作的方法为开发具有可控配位结构的高效SAC来增强碳氢化合物氧化反应提供了一定的理论支持和指导。
































 京公网安备 11010802027423号
京公网安备 11010802027423号