Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2023-12-22 , DOI: 10.1002/adsc.202301310 Juan Manuel Coto Cid 1 , Gonzalo de Gonzalo 2 , José A. Carmona 3 , Francisco Javier Iglesias Sigüenza 1 , Patricia Rodríguez Salamanca 4 , Rosario Fernández 2 , Valentín Hornillos 5 , José Lassaletta 6
|
Introduction
Axially chiral (hetero)biaryls, particularly those containing nitrogen atoms, are key structural motifs in several natural products, biologically active compounds, chiral ligands and catalysts.1 Several approaches have been established for the synthesis of biaryl compounds.2 However, methods detailing the preparation of heterobiaryl systems, especially (iso)quinoline derivatives, remain relatively limited.3 This scarcity becomes particularly evident when considering the synthesis of chiral 8-arylquinoline atropisomers, where a short handful of methods has been reported. Thus, in 2016, Zhou and coworkers described the kinetic resolution of 8-substituted quinolines by chiral phosphoric acid-catalysed asymmetric transfer hydrogenation (Scheme 1A).4 Later, the group of Miyaji described an enantioselective aromatic electrophilic halogenation reaction to afford chiral 8-arylquinoline derivatives, using a bifunctional Cinchona-derived organocatalyst (Scheme 1B).5 More recently, the groups of Shi6a and Zhu6b have reported Pd-catalysed cross-coupling strategies suitable for the synthesis of quinoline derivatives (Scheme 1C). Finally, axially chiral 8-arylquinoline derivatives have also been prepared by enantioselective C−H functionalization using a Pd/chiral phosphoric acid as a catalyst (Scheme 1D).3f, 7

Catalytic enantioselective synthesis of axially chiral quinolines. EHE: Diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate. CPA: Chiral phosphoric acid. STRIP: [6,6′-bis(2,4,6-triisopropylphenyl)-1,1-spirobiindan-7,7-diyl hydrogen phosphate.
Over the past few years, our group has successfully showcased the utilization of transient Lewis pairs (LABI) formed through the subtle interplay between an acidic moiety (even as weak as a carbonyl group) and a basic counterpart. This innovative approach has proven to be highly effective in promoting the racemization of stereogenic axes in heterobiaryl systems. The formation of quasi-zwitterionic transition states plays a pivotal role in facilitating racemization by substantially reducing the barrier to atropisomerization, which would otherwise be significantly higher in the absence of the stabilizing LABI interactions. In this scenario, we designed transformations aimed at eliminating the acidic nature of the carbonyl group through quaternization, thereby raising the barrier to atropisomerization, ultimately leading to configurationally stable enantioenriched compounds.
We first implemented this strategy in the atroposelective synthesis of chiral heterobiaryl carbinols through an asymmetric Zn-catalysed hydrosilylation of configurationally labile heterobiaryl ketones.8 The racemization of the substrate relied on a LABI interaction between the isoquinolyl nitrogen atom and the carbonyl group through the formation of 5-membered cyclic transition states. Furthermore, we developed the DKR of N- and 3-arylindole aldehydes that proceed by atroposelective bioreduction,9 and Rh-catalysed intermolecular reductive aldol reaction,10 respectively. In this case, racemization is expedited through a transient Lewis pair interaction occurring within a 6-membered cyclic transition state. The key factor for an efficient interaction was the presence of a five-membered heterocycle within the indole fragment that effectively elongates the distances between the Lewis base (NMe2 or SR) and the carbonyl fragment. This adjustment served to mitigate repulsive interactions between them, thus optimizing the overall process. More recently, the DKR of axially chiral 2-(quinolin-8-yl)benzaldehydes has been developed using Ir-catalysed transfer hydrogenative coupling with high yields and diastereoselectivities, and excellent levels of enantioselectivity (Scheme 1E).11 The racemization, facilitated by a LABI between the nitrogen in the quinoline and the aldehyde group, becomes more challenging in this case, since the cyclic structure enclosing the bonding interaction and both aryl rings are all six-membered.
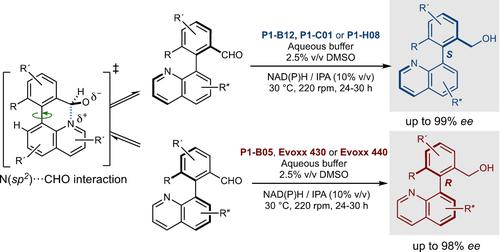
Given the prevalence of axially chiral 8-arylquinolines in a wide range of bioactive compounds, natural products,12 and ligands,13 the advancement of more effective methods for their synthesis is of great importance and continues to be a significant research goal. Therefore, in this study, we have employed our LABI strategy to synthesize a range of chiral 2-(quinolin-8-yl)benzyl alcohols via a dynamic kinetic resolution (DKR) catalysed by commercially available ketoreductases under mild reaction conditions (KREDs, Scheme 1E).14 It is worth noting that achieving high enantioselectivity in this scenario presents a greater challenge, as there is only a single stereogenic axis contributing to the overall stereoselectivity.
中文翻译:
生物催化动态动力学解析法选择性合成 2-(喹啉-8-基)苯甲醇
介绍
轴向手性(杂)联芳基,特别是那些含有氮原子的联芳基,是多种天然产物、生物活性化合物、手性配体和催化剂中的关键结构基序。1已经建立了几种联芳基化合物的合成方法。2然而,详细描述杂联芳体系,特别是(异)喹啉衍生物的制备方法仍然相对有限。3当考虑手性 8-芳基喹啉阻转异构体的合成时,这种稀缺性变得尤其明显,其中报道的方法很少。因此,2016年,Zhou和同事描述了通过手性磷酸催化的不对称转移氢化反应动力学拆分8-取代喹啉(方案1A)。4后来,Miyaji 团队描述了一种对映选择性芳香族亲电卤化反应,使用双功能金鸡纳衍生有机催化剂得到手性 8-芳基喹啉衍生物(方案 1B)。5最近,Shi 6a和 Zhu 6b小组报道了适用于合成喹啉衍生物的 Pd 催化交叉偶联策略(方案 1C)。最后,使用 Pd/手性磷酸作为催化剂,通过对映选择性 CH 官能化制备了轴向手性 8-芳基喹啉衍生物(方案 1D)。3楼, 7
轴向手性喹啉的催化对映选择性合成。EHE:2,6-二甲基-1,4-二氢吡啶-3,5-二甲酸二乙酯。CPA:手性磷酸。条带:[6,6′-双(2,4,6-三异丙基苯基)-1,1-螺二茚满-7,7-二基磷酸氢盐。
在过去的几年中,我们的团队成功地展示了通过酸性部分(甚至弱如羰基)和碱性对应物之间微妙的相互作用形成的瞬态路易斯对(LABI)的利用。这种创新方法已被证明在促进杂二芳基系统中立体轴的外消旋化方面非常有效。准两性离子过渡态的形成通过大幅降低阻转异构化的障碍而在促进外消旋化方面发挥着关键作用,否则在没有稳定的 LABI 相互作用的情况下,阻转异构化的障碍会显着更高。在这种情况下,我们设计了旨在通过季铵化消除羰基的酸性性质的转化,从而提高阻转异构化的障碍,最终产生构型稳定的对映体富集化合物。
我们首先通过构型不稳定的杂二芳基酮的不对称锌催化氢化硅烷化,在手性杂二芳基甲醇的逆向选择性合成中实施了这一策略。8底物的外消旋作用依赖于异喹啉基氮原子和羰基之间通过形成 5 元环状过渡态的 LABI 相互作用。此外,我们还开发了 N- 和 3- 芳基吲哚醛的 DKR,分别通过天体选择性生物还原9和 Rh 催化的分子间还原羟醛反应10进行。在这种情况下,通过六元循环过渡态内发生的瞬时路易斯对相互作用加速外消旋化。有效相互作用的关键因素是吲哚片段中存在五元杂环,它有效地延长了路易斯碱(NMe 2或 SR)和羰基片段之间的距离。这种调整有助于减轻它们之间的排斥相互作用,从而优化整个过程。最近,使用 Ir 催化的转移氢化偶联开发了轴向手性 2-(喹啉-8-基)苯甲醛的 DKR,具有高产率和非对映选择性,以及出色的对映选择性水平(方案 1E)。11在这种情况下,喹啉中的氮和醛基之间的 LABI 促进的外消旋化变得更具挑战性,因为封闭键合相互作用的环状结构和两个芳基环都是六元的。
鉴于轴向手性 8-芳基喹啉在各种生物活性化合物、天然产物12和配体13中普遍存在,开发更有效的合成方法非常重要,并且仍然是一个重要的研究目标。因此,在本研究中,我们采用 LABI 策略,通过市售酮还原酶在温和反应条件下催化的动态动力学拆分 (DKR) 合成了一系列手性 2-(喹啉-8-基) 苯甲醇(KREDs,方案) 1E)。14值得注意的是,在这种情况下实现高对映选择性提出了更大的挑战,因为只有一个立体轴对整体立体选择性有贡献。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号