Acta Neuropathologica ( IF 9.3 ) Pub Date : 2023-12-11 , DOI: 10.1007/s00401-023-02654-1 Sara Hadad 1 , Rohit Gupta 2 , Nancy Ann Oberheim Bush 1, 3 , Jennie W Taylor 1, 3 , Javier E Villanueva-Meyer 4 , Jacob S Young 1 , Jasper Wu 2 , Ajay Ravindranathan 2 , Yalan Zhang 1 , Gayathri Warrier 1 , Lucie McCoy 1 , Anny Shai 1 , Melike Pekmezci 2 , Arie Perry 1, 2 , Andrew W Bollen 2 , Joanna J Phillips 1, 2 , Steve E Braunstein 5 , David R Raleigh 1, 5 , Philip Theodosopoulos 1 , Manish K Aghi 1 , Edward F Chang 1 , Shawn L Hervey-Jumper 1 , Joseph F Costello 1 , John de Groot 1 , Nicholas A Butowski 1 , Jennifer L Clarke 1, 3 , Susan M Chang 1 , Mitchel S Berger 1 , Annette M Molinaro 1 , David A Solomon 2
|
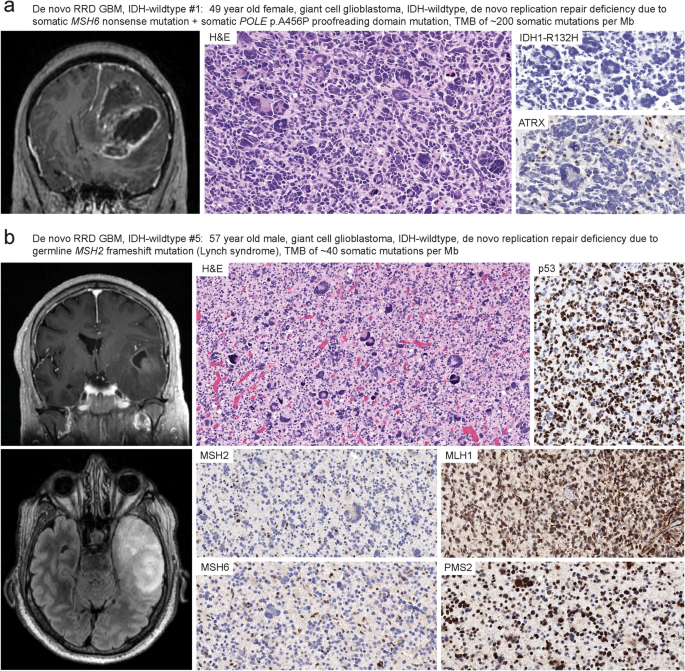
Glioblastoma is a clinically and molecularly heterogeneous disease, and new predictive biomarkers are needed to identify those patients most likely to respond to specific treatments. Through prospective genomic profiling of 459 consecutive primary treatment-naïve IDH-wildtype glioblastomas in adults, we identified a unique subgroup (2%, 9/459) defined by somatic hypermutation and DNA replication repair deficiency due to biallelic inactivation of a canonical mismatch repair gene. The deleterious mutations in mismatch repair genes were often present in the germline in the heterozygous state with somatic inactivation of the remaining allele, consistent with glioblastomas arising due to underlying Lynch syndrome. A subset of tumors had accompanying proofreading domain mutations in the DNA polymerase POLE and resultant “ultrahypermutation”. The median age at diagnosis was 50 years (range 27–78), compared with 63 years for the other 450 patients with conventional glioblastoma (p < 0.01). All tumors had histologic features of the giant cell variant of glioblastoma. They lacked EGFR amplification, lacked combined trisomy of chromosome 7 plus monosomy of chromosome 10, and only rarely had TERT promoter mutation or CDKN2A homozygous deletion, which are hallmarks of conventional IDH-wildtype glioblastoma. Instead, they harbored frequent inactivating mutations in TP53, NF1, PTEN, ATRX, and SETD2 and recurrent activating mutations in PDGFRA. DNA methylation profiling revealed they did not align with known reference adult glioblastoma methylation classes, but instead had unique globally hypomethylated epigenomes and mostly classified as “Diffuse pediatric-type high grade glioma, RTK1 subtype, subclass A”. Five patients were treated with immune checkpoint blockade, four of whom survived greater than 3 years. The median overall survival was 36.8 months, compared to 15.5 months for the other 450 patients (p < 0.001). We conclude that “De novo replication repair deficient glioblastoma, IDH-wildtype” represents a biologically distinct subtype in the adult population that may benefit from prospective identification and treatment with immune checkpoint blockade.
中文翻译:
“De novo replication repair deficient glioblastoma, IDH 野生型”是成人中一种独特的胶质母细胞瘤亚型,可能受益于免疫检查点阻断
胶质母细胞瘤是一种临床和分子异质性疾病,需要新的预测性生物标志物来识别最有可能对特定治疗产生反应的患者。通过对 459 例连续的成人初治原发性 IDH 野生型胶质母细胞瘤进行前瞻性基因组分析,我们确定了一个独特的亚组 (2%,9/459),定义为由于经典错配修复基因的双等位基因失活而导致的体细胞超突变和 DNA 复制修复缺陷。错配修复基因的有害突变通常以杂合状态存在于种系中,剩余等位基因体细胞失活,与潜在 Lynch 综合征引起的胶质母细胞瘤一致。一部分肿瘤在 DNA 聚合酶 POLE 中伴有校对结构域突变,并导致“超超突变”。诊断时的中位年龄为 50 岁 (范围 27-78),而其他 450 名常规胶质母细胞瘤患者为 63 岁 (p < 0.01)。所有肿瘤均具有胶质母细胞瘤巨细胞变异的组织学特征。他们缺乏 EGFR 扩增,缺乏 7 号染色体三体和 10 号染色体单体的联合三体,并且很少出现 TERT 启动子突变或 CDKN2A 纯合缺失,这是传统 IDH 野生型胶质母细胞瘤的标志。相反,它们在 TP53 、 NF1 、 PTEN、 ATRX 和 SETD2 中具有频繁的失活突变,并且在 PDGFRA 中具有复发性的激活突变。 DNA 甲基化分析显示它们与已知的参考成人胶质母细胞瘤甲基化类别不一致,而是具有独特的全球低甲基化表观基因组,并且大多归类为“弥漫性儿科型高级别胶质瘤,RTK1 亚型,亚类 A”。5 例患者接受了免疫检查点阻断治疗,其中 4 例存活时间超过 3 年。中位总生存期为 36.8 个月,而其他 450 例患者的中位总生存期为 15.5 个月 (p < 0.001)。我们得出结论,“De novo replication repair deficient glioblastoma, IDH 野生型”代表了成人人群中生物学上不同的亚型,可能受益于免疫检查点阻断的前瞻性识别和治疗。
































 京公网安备 11010802027423号
京公网安备 11010802027423号