当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Heptazine, Cyclazine, and Related Compounds: Chemically-Accurate Estimates of the Inverted Singlet–Triplet Gap
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2023-12-04 , DOI: 10.1021/acs.jpclett.3c03042 Pierre-François Loos 1 , Filippo Lipparini 2 , Denis Jacquemin 3, 4
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2023-12-04 , DOI: 10.1021/acs.jpclett.3c03042 Pierre-François Loos 1 , Filippo Lipparini 2 , Denis Jacquemin 3, 4
Affiliation
|
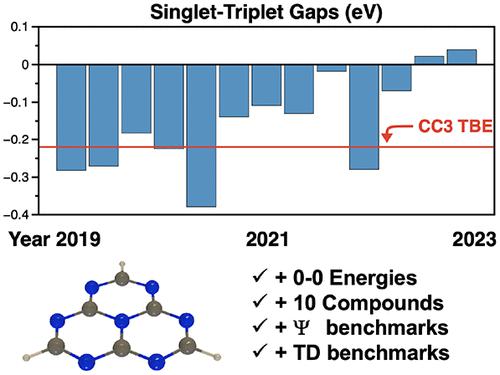
Molecules that violate Hund’s rule and exhibit an inverted gap between the lowest singlet S1 and triplet T1 excited states have attracted considerable attention due to their potential applications in optoelectronics. Among these molecules, the triangular-shaped heptazine, and its derivatives, have been in the limelight. However, conflicting reports have arisen regarding the relative energies of S1 and T1. Here, we employ highly accurate levels of theory, such as CC3, to not only resolve the debate concerning the sign but also quantify the magnitude of the S1–T1 gap. We also determined the 0–0 energies to evaluate the significance of the vertical approximation. In addition, we compute reference S1–T1 gaps for a series of 10 related molecules. This enables us to benchmark lower-order methods for future applications in larger systems within the same family of compounds. This contribution can serve as a foundation for the design of triangular-shaped molecules with enhanced photophysical properties.
中文翻译:

庚嗪、环嗪和相关化合物:反向单线态-三线态间隙的化学精确估计
违反洪德规则并在最低单线态S 1和三线态T 1激发态之间表现出倒置能隙的分子由于其在光电子学中的潜在应用而引起了相当大的关注。在这些分子中,三角形的庚嗪及其衍生物备受关注。然而,关于S 1和T 1的相对能量出现了相互矛盾的报告。在这里,我们采用高度准确的理论水平,例如 CC3,不仅可以解决有关符号的争论,还可以量化 S 1 –T 1间隙的大小。我们还确定了 0-0 能量来评估垂直近似的显着性。此外,我们计算了一系列 10 个相关分子的参考 S 1 –T 1间隙。这使我们能够对低阶方法进行基准测试,以便将来在同一化合物系列的较大系统中应用。这一贡献可以作为设计具有增强光物理性质的三角形分子的基础。
更新日期:2023-12-04
中文翻译:
庚嗪、环嗪和相关化合物:反向单线态-三线态间隙的化学精确估计
违反洪德规则并在最低单线态S 1和三线态T 1激发态之间表现出倒置能隙的分子由于其在光电子学中的潜在应用而引起了相当大的关注。在这些分子中,三角形的庚嗪及其衍生物备受关注。然而,关于S 1和T 1的相对能量出现了相互矛盾的报告。在这里,我们采用高度准确的理论水平,例如 CC3,不仅可以解决有关符号的争论,还可以量化 S 1 –T 1间隙的大小。我们还确定了 0-0 能量来评估垂直近似的显着性。此外,我们计算了一系列 10 个相关分子的参考 S 1 –T 1间隙。这使我们能够对低阶方法进行基准测试,以便将来在同一化合物系列的较大系统中应用。这一贡献可以作为设计具有增强光物理性质的三角形分子的基础。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号