Nature Communications ( IF 14.7 ) Pub Date : 2023-12-01 , DOI: 10.1038/s41467-023-43600-9 Zixiang Zhou 1, 2 , Yunshan Zhong 1 , Zemin Zhang 1, 2 , Xianwen Ren 1
|
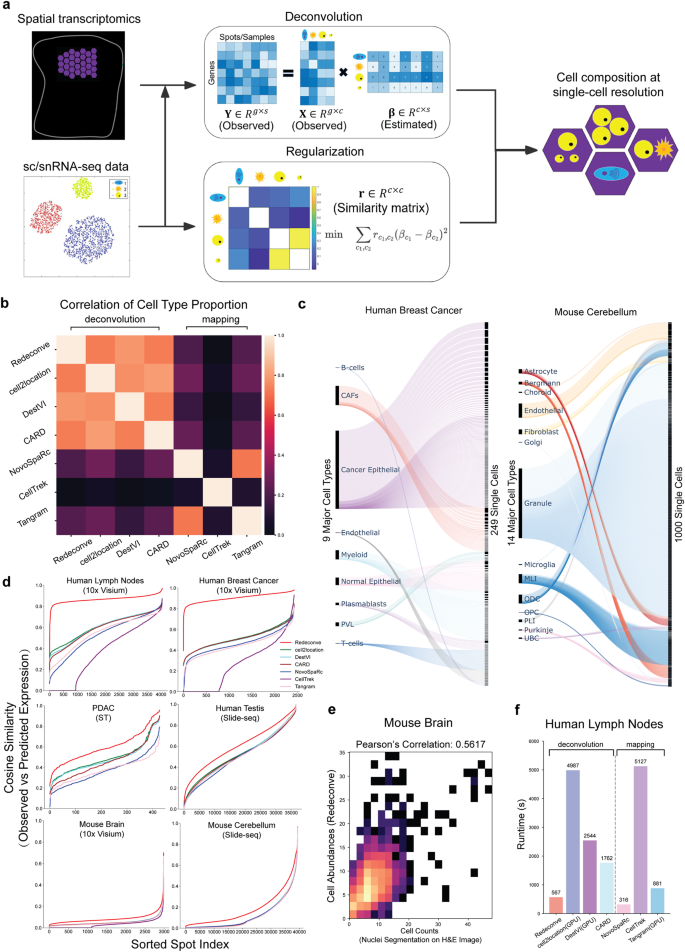
Computational deconvolution with single-cell RNA sequencing data as reference is pivotal to interpreting spatial transcriptomics data, but the current methods are limited to cell-type resolution. Here we present Redeconve, an algorithm to deconvolute spatial transcriptomics data at single-cell resolution, enabling interpretation of spatial transcriptomics data with thousands of nuanced cell states. We benchmark Redeconve with the state-of-the-art algorithms on diverse spatial transcriptomics platforms and datasets and demonstrate the superiority of Redeconve in terms of accuracy, resolution, robustness, and speed. Application to a human pancreatic cancer dataset reveals cancer-clone-specific T cell infiltration, and application to lymph node samples identifies differential cytotoxic T cells between IgA+ and IgG+ spots, providing novel insights into tumor immunology and the regulatory mechanisms underlying antibody class switch.
中文翻译:
使用 Redeconve 以单细胞分辨率进行空间转录组解卷积
以单细胞 RNA 测序数据为参考的计算反卷积对于解释空间转录组数据至关重要,但目前的方法仅限于细胞类型分辨率。在这里,我们推出了 Redeconve,一种以单细胞分辨率对空间转录组数据进行解卷积的算法,能够解释具有数千个细微细胞状态的空间转录组数据。我们在不同的空间转录组学平台和数据集上使用最先进的算法对 Redeconve 进行基准测试,并证明了 Redeconve 在准确性、分辨率、鲁棒性和速度方面的优越性。对人类胰腺癌数据集的应用揭示了癌症克隆特异性 T 细胞浸润,对淋巴结样本的应用识别了 IgA+ 和 IgG+ 点之间的差异细胞毒性 T 细胞,为肿瘤免疫学和抗体类别转换背后的调节机制提供了新的见解。






























 京公网安备 11010802027423号
京公网安备 11010802027423号