Nature Communications ( IF 14.7 ) Pub Date : 2023-12-01 , DOI: 10.1038/s41467-023-43496-5 Eli Gerber 1 , Steven B Torrisi 2, 3 , Sara Shabani 4 , Eric Seewald 4 , Jordan Pack 4 , Jennifer E Hoffman 2, 5 , Cory R Dean 4 , Abhay N Pasupathy 4 , Eun-Ah Kim 6
|
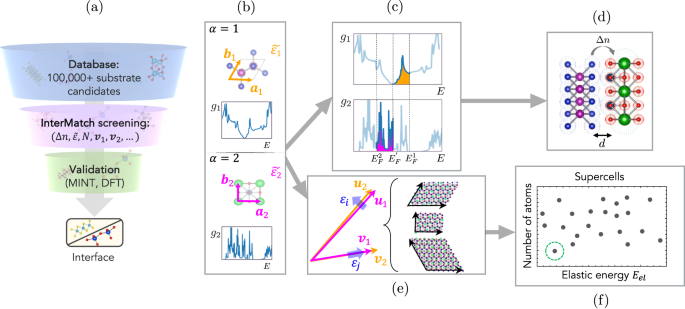
Forming a hetero-interface is a materials-design strategy that can access an astronomically large phase space. However, the immense phase space necessitates a high-throughput approach for an optimal interface design. Here we introduce a high-throughput computational framework, InterMatch, for efficiently predicting charge transfer, strain, and superlattice structure of an interface by leveraging the databases of individual bulk materials. Specifically, the algorithm reads in the lattice vectors, density of states, and the stiffness tensors for each material in their isolated form from the Materials Project. From these bulk properties, InterMatch estimates the interfacial properties. We benchmark InterMatch predictions for the charge transfer against experimental measurements and supercell density-functional theory calculations. We then use InterMatch to predict promising interface candidates for doping transition metal dichalcogenide MoSe2. Finally, we explain experimental observation of factor of 10 variation in the supercell periodicity within a few microns in graphene/α-RuCl3 by exploring low energy superlattice structures as a function of twist angle using InterMatch. We anticipate our open-source InterMatch algorithm accelerating and guiding ever-growing interfacial design efforts. Moreover, the interface database resulting from the InterMatch searches presented in this paper can be readily accessed online.
中文翻译:
使用 InterMatch 进行原子接口的高通量从头设计
形成异质界面是一种材料设计策略,可以访问天文数字般大的相空间。然而,巨大的相空间需要采用高通量方法来实现最佳界面设计。在这里,我们介绍了一个高通量计算框架 InterMatch,用于通过利用单个散装材料的数据库来有效预测界面的电荷转移、应变和超晶格结构。具体来说,该算法从材料项目中以孤立的形式读取每种材料的晶格向量、态密度和刚度张量。 InterMatch 根据这些整体特性估算界面特性。我们根据实验测量和超级单元密度泛函理论计算对 InterMatch 的电荷转移预测进行基准测试。然后,我们使用 InterMatch 来预测有希望的掺杂过渡金属二硫族化物 MoSe 2的候选界面。最后,我们通过使用 InterMatch 探索低能超晶格结构作为扭转角的函数,解释了在石墨烯/ α -RuCl 3中几微米内超级晶胞周期性发生 10 倍变化的实验观察结果。我们预计我们的开源 InterMatch 算法将加速并指导不断发展的界面设计工作。此外,本文提出的 InterMatch 搜索产生的界面数据库可以轻松在线访问。










































 京公网安备 11010802027423号
京公网安备 11010802027423号