Child's Nervous System ( IF 1.3 ) Pub Date : 2023-11-23 , DOI: 10.1007/s00381-023-06232-4 Alice Antico 1, 2 , Francesca Vitulli 1, 3 , Andrea Rossi 4, 5 , Gabriele Gaggero 6 , Gianluca Piatelli 1 , Alessandro Consales 1
|
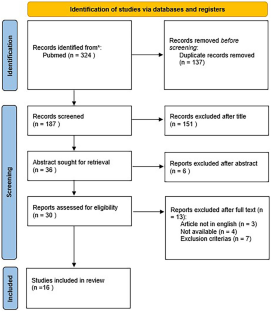
Autosomal dominantly inherited neurofibromatosis type I (NF1) is a systemic disorder caused by a mutation of a gene on chromosome 17q11.2 and characterized by multiple café-au-lait spots, lentiginous macules, Lisch nodules of the iris, and tumors of the nervous system. Bony manifestations such as scoliosis, dysplasia of the greater sphenoidal wing, tibial pseudoarthrosis, short stature, and macrocephaly have been reported in approximately 50% of patients. However, calvarial bone defects are rare. After screening 324 articles, 23 cases (12 adult and 11 pediatric patients) of occipital bone defects in NF1 patients were selected. All patients had a single/multiple bone defect over the lambdoid suture. Adjacent benign plexiform neurofibromas were observed in 14 patients (60.8%, 7 adults and 7 children); one adult patient was diagnosed with neurofibrosarcoma. Meningoencephalocele over the occipital defect was noted in 8 cases (34.78%, all adults). Cranioplasty was performed in only 17.39% of patients. Histologic examination was performed in 7 of the 15 patients with associated neurofibromas/neurofibrosarcomas. Biopsy of the bone margins surrounding the defect was performed in only one case. Pathologic examination of the herniated parieto-occipital or cerebellar tissue was not performed in any of the patients studied. We report the case of a 9-year-old girl with NF1 and a significant occipital bone defect and performed a systematic review of the relevant literature to highlight the challenges in treating this condition and to investigate the underlying mechanisms contributing to bone defects or dysplasia in NF1.
中文翻译:
NF1儿童巨大枕骨缺损合并脑膜脑膨出的发病机制及治疗:病例报告及文献复习
常染色体显性遗传 I 型神经纤维瘤病 (NF1) 是一种由染色体 17q11.2 上的基因突变引起的系统性疾病,其特征为多发咖啡斑、雀斑、虹膜 Lisch 结节和神经肿瘤系统。据报道,约 50% 的患者出现脊柱侧弯、蝶骨大翼发育不良、胫骨假关节、身材矮小和巨头畸形等骨表现。然而,颅骨骨缺损很少见。经过筛选324篇文章,筛选出23例(12例成人和11例儿童)枕骨缺损的NF1患者。所有患者的人字形缝合处均存在单/多发骨缺损。 14 名患者(60.8%,7 名成人和 7 名儿童)观察到邻近良性丛状神经纤维瘤;一名成年患者被诊断患有神经纤维肉瘤。枕骨缺损处出现脑膜脑膨出 8 例(34.78%,均为成人)。仅 17.39% 的患者进行了颅骨成形术。对 15 名患有相关神经纤维瘤/神经纤维肉瘤的患者中的 7 名进行了组织学检查。仅在 1 例病例中对缺损周围的骨边缘进行了活检。没有对所有研究的患者进行顶枕或小脑组织疝出的病理检查。我们报告了一名 9 岁女孩患有 NF1 和枕骨骨缺损的病例,并对相关文献进行了系统回顾,以强调治疗这种疾病的挑战,并调查导致骨缺损或发育不良的潜在机制。 NF1。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号