当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effect of Co/SiO2 Single-Site Heterogeneity on Small Alkane Dehydrogenation Kinetics
ACS Catalysis ( IF 11.3 ) Pub Date : 2023-11-20 , DOI: 10.1021/acscatal.3c03273
Sanjana Srinivas 1, 2 , Kaveri Srivastava 1 , Kewei Yu 1, 2 , Dionisios G. Vlachos 1, 2 , Stavros Caratzoulas 2
ACS Catalysis ( IF 11.3 ) Pub Date : 2023-11-20 , DOI: 10.1021/acscatal.3c03273
Sanjana Srinivas 1, 2 , Kaveri Srivastava 1 , Kewei Yu 1, 2 , Dionisios G. Vlachos 1, 2 , Stavros Caratzoulas 2
Affiliation
|
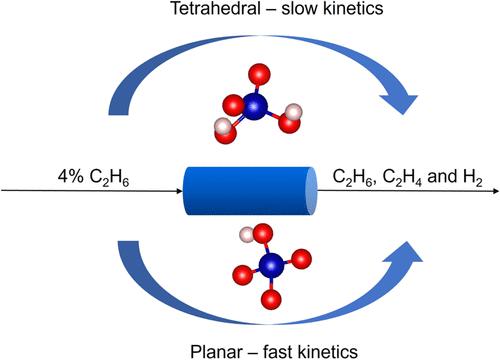
Atomically dispersed, high-spin Co(II) atoms in distorted tetrahedral coordination to an amorphous silica (am-SiO2) support and more recently in zeolite frameworks are active and selective for light alkane dehydrogenation. This article investigates how variations in the geometry of the active sites affect the ethane dehydrogenation activity of atomically dispersed Co(II) on an am-SiO2 support. We generate a distribution of sites and determine the geometric parameters that exhibit the strongest correlation with the coordination geometry and activity of the metal atom by means of linear dimensionality reduction techniques. We perform electronic structure calculations and microkinetic modeling and deduce the mechanism and kinetics for a representative sample of sites. Irrespective of the active site geometry, the rate of ethane dehydrogenation is governed by the β-hydride elimination, which involves quartet-doublet spin-crossing and proceeds adiabatically due to strong spin–orbit coupling. Informed by the complete microkinetic analysis of the sites, we derive the reduced rate expression as a function of three site-dependent quantities. We show that these site-dependent quantities correlate with the energy of formation of the ethyl intermediate that forms via C–H bond activation. This correlation allows us to derive the site-averaged rate for the entire distribution of sites. Among various sites, the tricoordinate and planar tetra-coordinate Co sites exhibit higher Lewis acidity than the tetrahedral sites, and consequently, higher initial rates. We discuss the implications for more active catalysts.
中文翻译:

Co/SiO2单中心异质性对小烷烃脱氢动力学的影响
原子分散的高自旋 Co(II) 原子以扭曲四面体配位到无定形二氧化硅 (am-SiO 2 ) 载体上,并且最近在沸石骨架中,对于轻质烷烃脱氢具有活性和选择性。本文研究了活性位点几何形状的变化如何影响 am-SiO 2载体上原子分散的 Co(II) 的乙烷脱氢活性。我们生成位点分布,并通过线性降维技术确定与金属原子的配位几何和活性最强相关的几何参数。我们进行电子结构计算和微动力学建模,并推断出代表性位点样本的机制和动力学。无论活性位点的几何形状如何,乙烷脱氢的速率均受β-氢化物消除的控制,其中涉及四重态-双重态自旋交叉,并由于强自旋轨道耦合而绝热进行。根据位点的完整微动力学分析,我们推导出降低的速率表达作为三个位点相关量的函数。我们表明,这些位点相关的量与通过 C-H 键激活形成的乙基中间体的形成能量相关。这种相关性使我们能够得出整个站点分布的站点平均速率。在各种位点中,三配位和平面四配位Co位点比四面体位点表现出更高的路易斯酸性,因此具有更高的初始速率。我们讨论了对更活跃的催化剂的影响。
更新日期:2023-11-20
中文翻译:
Co/SiO2单中心异质性对小烷烃脱氢动力学的影响
原子分散的高自旋 Co(II) 原子以扭曲四面体配位到无定形二氧化硅 (am-SiO 2 ) 载体上,并且最近在沸石骨架中,对于轻质烷烃脱氢具有活性和选择性。本文研究了活性位点几何形状的变化如何影响 am-SiO 2载体上原子分散的 Co(II) 的乙烷脱氢活性。我们生成位点分布,并通过线性降维技术确定与金属原子的配位几何和活性最强相关的几何参数。我们进行电子结构计算和微动力学建模,并推断出代表性位点样本的机制和动力学。无论活性位点的几何形状如何,乙烷脱氢的速率均受β-氢化物消除的控制,其中涉及四重态-双重态自旋交叉,并由于强自旋轨道耦合而绝热进行。根据位点的完整微动力学分析,我们推导出降低的速率表达作为三个位点相关量的函数。我们表明,这些位点相关的量与通过 C-H 键激活形成的乙基中间体的形成能量相关。这种相关性使我们能够得出整个站点分布的站点平均速率。在各种位点中,三配位和平面四配位Co位点比四面体位点表现出更高的路易斯酸性,因此具有更高的初始速率。我们讨论了对更活跃的催化剂的影响。
































 京公网安备 11010802027423号
京公网安备 11010802027423号