Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2023-11-22 , DOI: 10.1002/adsc.202301116
Alexandre Bergounioux 1 , Romane Lhotellier 1 , Thierry Roisnel 2 , Nicolas GOUAULT 3 , Gilles Argouarch 4 , Claudia LALLI 1
|
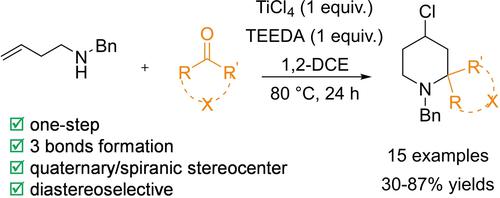
The modern Prins cyclization involves simple starting materials such as a homoallylic alcohol or amine and an aldehyde, to provide tetrahydropyrans or piperidines in only one step with the concomitant carbon-carbon and carbon-heteroatom bond formation. While the Prins cyclization is very well documented,1 its nitrogen-based version is significantly less developed,2 although the piperidine ring is more widespread than tetrahydropyrans in natural products and drugs.3 Despite the great potential of aza-Prins cyclization for the synthesis of natural and/or bioactive compounds, particular attention has been paid solely to the diversification of the homoallylic amines. In particular, aza-silyl Prins reactions4 have been developed to obtain 3,4-dehydropiperidines with loss of the silicon group, alkynyl aza-Prins annulations5 have provided similar compounds with a nucleophile in position C-4, and finally cascade aza-Prins reactions6 have been reported for the formation of fused bi- and tricyclic systems. Concerning the carbonyl partner, apart from aldehydes, only two examples have been published to date: one with a very narrow range of ketones7 and the other limited to 1,2-dicarbonyl substrates,8 both leading to the synthesis of piperidines containing a tetrasubstituted carbon stereocenter in position 2.
To the best of our knowledge, an effective aza-Prins cyclization with non-activated ketones has not been reported so far. In continuation with our interest in Prins processes,9 we disclose here the first aza-Prins cyclization with aliphatic, aromatic, and heterocyclic ketones. Noteworthy, the present method competes well with other accesses to 2-spiranic piperidines.10
We have already reported the effectiveness of a synergistic combination of Lewis and Brønsted acids in the aza-Prins cyclization between secondary alkyl homoallylic amines and aldehydes.11 Therefore, we first investigated the reactivity between N-benzyl homoallylamine 1 and cyclohexanone (2 a) in the presence of 1 equiv. of TiCl4 and 0.1 equiv. of p-TsOH ⋅ H2O in DCM at 60 °C in a sealed Schlenk tube for 24 h (Table 1, entry 1). Unfortunately, a disappointing lack of reactivity has been observed. We next investigated the effect of the Brønsted and Lewis acids alone (Table 1, entries 2 and 3 respectively), and only 9% conversion of the desired 2-spiropiperidine 3 a could be observed in the presence of 1 equiv. of TiCl4 by 1H-NMR analysis of the crude reaction mixture. Encouraged by this result, we decided to explore the effect of different additives such as molecular sieves, water and also PPh3 (Table 1, entries 4–6) without further improvements. Gratifyingly, the use of 1 and then of 2 equiv. of triethylamine (TEA) (Table 1, entries 7 and 8) led to respectively 25% and 52% conversion into the desired product. This last result prompted us to examine diamines, in particular N,N,N’,N’-tetramethylethylenediamine (TMEDA), N,N,N’,N’-tetraethylethylenediamine (TEEDA), 1,4-diazabicyclo2,2,2octane (DABCO), and 1,2-dipiperidinylethane. As depicted in entries 9–12 in Table 1, TEEDA was found to be superior, leading to 2-spiropiperidine 3 a in 61% conversion, and was retained as the additive of choice for the rest of this study. In order to further improve the reactivity, the reaction was next performed in 1,2-DCE at 80 °C and indeed the analysis of the crude mixture showed 75% conversion (Table 1, entry 13). Increasing again the temperature to 100 °C proved detrimental to the reaction as by-products arising from the self-condensation/enolization of ketone 2 a became too abundant. Finally, the stepwise addition of an excess of ketone during the reaction, i. e. 1.5 equiv. at the start followed by 0.5 equiv. after 8 hours, allowed us to obtain 3 a in 90% conversion (Table 1, entry 14).
|
||||
Entry[a] |
Additive (equiv.) |
Solvent |
T (°C) |
Conv. (%)[b] |
|---|---|---|---|---|
1 |
p-TsOH ⋅ H2O (0.1) |
DCM |
60 |
0 |
2[c] |
p-TsOH ⋅ H2O (1) |
DCM |
60 |
0 |
3 |
none |
DCM |
60 |
9 |
4 |
MS (3 Å) |
DCM |
60 |
0 |
5 |
H2O (1) |
DCM |
60 |
0 |
6 |
PPh3 (1) |
DCM |
60 |
0 |
7 |
TEA (1) |
DCM |
60 |
25 |
8 |
TEA (2) |
DCM |
60 |
52 |
9 |
TMEDA (1) |
DCM |
60 |
58 |
10 |
TEEDA (1) |
DCM |
60 |
61 |
11 |
DABCO (1) |
DCM |
60 |
43 |
12 |
1,2-dipiperidinyl ethane (1) |
DCM |
60 |
55 |
13 |
TEEDA (1) |
1,2-DCE |
80 |
75 |
14[d] |
TEEDA (1) |
1,2-DCE |
80 |
90 |

- [a] Unless otherwise noted, reactions were performed with homoallylamine 1 (1 mmol, 1 equiv.), 2 a (1 mmol, 1 equiv.), and TiCl4 (1 mmol, 1 equiv.) in a sealed Schlenk tube for 24 h. [b] Conversions were calculated by 1H-NMR analysis of the crude reaction mixtures. [c] Reaction was performed without TiCl4. [d] Reaction was performed with 1.5+0.5 equiv. of 2 a.
Under the above optimized conditions, compound 3 a was isolated in 63% yield and the scope of the reaction was first evaluated on various other cyclic ketones (Scheme 1). Cyclopentanone (2 b) and cycloheptanone (2 c) gave satisfactory results albeit both reaction needed to be carried out at 100 °C, and products 3 b and 3 c were isolated in 30% and 49% yields, respectively. Next, a series of cyclohexanone derivatives substituted at the C-4 position 2 d–f were screened, and regardless the presence of an alkyl or aryl group in this position, these substrates readily participated in the reaction leading to 3 d, 3 e, and 3 f in 87%, 57%, and 58% yields, respectively. It has to be stressed that cyclanic isomerism has been observed for this series of 2-spiropiperidines and that the formation of the most stable trans isomer is favoured, if not exclusive, as in the case of 3 e possessing the bulky t-Bu group. When present, the two isomers are separable by column chromatography on silica gel and the relative stereochemistry of 3 f-trans has been unambiguously confirmed by single crystal X-ray analysis, as shown in Figure 1.12

Scope of the aza-Prins cyclization. Reaction conditions: 1 (1 mmol), 2 (1.5+0.5 equiv.), TiCl4 (1 equiv.), TEEDA (1 equiv.) in 1,2-DCE at 80 °C in a sealed Schlenk tube for 24 h. Yields were determined after purification by column chromatography on silica gel (General procedure A).[a] Reactions performed with 2 (1.5+1.5 equiv.) at 100 °C (General procedure B).[b] Reactions performed without TEEDA (General procedure C).

ORTEP representations of 3 f-trans (top) and 3 l-trans (bottom) at 50% thermal ellipsoids. Hydrogen atoms are omitted for clarity.
The aza-Prins cyclization was then applied to more challenging acyclic substrates. In the presence of 3-pentanone (2 g), 4-heptanone (2 h), and benzophenone (2 i), we were pleased to observe the formation of compounds 3 g–i. Given the difficulty of constructing tetrasubstituted carbon stereocenters from acyclic systems due to their inherent conformational mobility,13 the yields obtained here, ranging from 30% to 34%, are still acceptable.
Encouraged by those results, we next examined the reaction between N-benzyl homoallylamine 1 and various activated (di)ketones. We were satisfied to conclude that this panel efficiently underwent aza-Prins cyclization with a very marked diastereoselectivity in favour of the trans isomer. As depicted in Scheme 1, benzil (2 j) and its analog 4,4’-dimethylbenzil (2 k) gave separable mixtures of diastereomers 3 j and 3 k with overall yields of 64% and 50%, respectively. The use of keto-ester 2 l allowed to recover 3 l-trans and 3 l-cis in 70% yield, whereas trifluoromethylated ketone 2 m gave piperidines 3 m-trans and 3 m-cis in 60% overall yield. Moreover, single crystal X-ray analysis of 3 l-trans allowed to establish the relative stereochemistry of the two stereogenic centers that were formed during the reaction (Figure 1).12
Finally, we looked at the reactivity of N-heterocyclic ketones. Piperidones in particular were considered because their reaction could provide an access to spiro-fused dipiperidines, a class of compounds withacc strong biological activities.14 Therefore, the aza-Prins cyclization between 1 and 1-methyl-4-piperidone (2 n) was attempted under the optimized conditions but led to a loss of the reactivity. It was assumed that, if TEEDA is required to carry out the reaction, the combination of this additive and a nitrogen-containing substrate could deactivate the titanium active species. Indeed, without adding TEEDA to the reaction medium, the reactivity was recovered and product 3 n was isolated in 60% yield (Scheme 1). This trend could be extended to 1-benzyl-4-piperidone (2 o), leading to the corresponding dipiperidine 3 o in 49% yield.
In summary, we have developed a titanium-TEEDA promoted aza-Prins cyclization of aliphatic, aromatic, and heterocyclic ketones. Our methodology provides an access to piperidines bearing tetrasubstituted or even spiranic carbon centers at the C-2 position, which are common naturally occuring scaffolds. The reaction proceeds diastereoselectively in favour of the trans isomer, both when non-symmetric substrates are used and when cyclanic isomerism is present. Further applications and substrate variations are in progress in our laboratory.
Experimental Section
General Procedure A
To a solution of N-benzyl homoallylamine 1 (163 mL, 1 mmol) and the ketone (1.5 mmol) in dry 1,2-DCE (10 mL) was added TEEDA (213 mL, 1 mmol). The mixture was stirred for 10 min and a solution of TiCl4 (1 mL, 1.0 M in DCM, 1 mmol) was added dropwise. The solution was stirred at 80 °C for 8 hours. After cooling to room temperature, the ketone (0.5 mmol) was again added to the mixture and the solution was further stirred at 80 °C overnight. After cooling to room temperature, the reaction was quenched with sat. NaHCO3 (15 mL) and filtered on a Buchner funnel (1 layer of filter paper). The product was extracted with DCM (2×20 mL), the combined organic phases were washed with water (1×20 mL), dried over anhydrous MgSO4, and evaporated to dryness. The residue was purified by column chromatography on silica gel.
General Procedure B
To a solution of N-benzyl homoallylamine 1 (163 mL, 1 mmol) and the ketone (1.5 mmol) in dry 1,2-DCE (10 mL) was added TEEDA (213 mL, 1 mmol). The mixture was stirred for 10 min and a solution of TiCl4 (1 mL, 1.0 M in DCM, 1 mmol) was added dropwise. The solution was stirred at 100 °C for 8 hours. After cooling to room temperature, the ketone (1.5 mmol) was again added to the mixture and the solution was further stirred at 100 °C overnight. After cooling to room temperature, the reaction was quenched with sat. NaHCO3 (15 mL) and filtered on a Buchner funnel (1 layer of filter paper). The product was extracted with DCM (2×20 mL), the combined organic phases were washed with water (1×20 mL), dried over anhydrous MgSO4, and evaporated to dryness. The residue was purified by column chromatography on silica gel.
General Procedure C
A solution of N-benzyl homoallylamine 1 (163 mL, 1 mmol) and the ketone (1.5 mmol) in dry 1,2-DCE (10 mL) was stirred for 10 min before adding a solution of TiCl4 (1 mL, 1.0 M in DCM, 1 mmol) dropwise. The solution was stirred at 80 °C for 8 hours. After cooling to room temperature, the ketone (0.5 mmol) was again added to the mixture and the solution was further stirred at 80 °C overnight. After cooling to room temperature, the reaction was quenched with sat. NaHCO3 (15 mL) and filtered on a Buchner funnel (1 layer of filter paper). The product was extracted with DCM (2×20 mL), the combined organic phases were washed with water (1×20 mL), dried over anhydrous MgSO4, and evaporated to dryness. The residue was purified by column chromatography on silica gel.
Acknowledgments
The authors thank Université de Rennes and CNRS for financial support. The CRMPO of the Institut des Sciences Chimiques de Rennes for mass measurement, the Centre de Diffractométrie X for X-ray analyses and FEDER funds for acquisition of single-crystal equipment are gratefully acknowledged. Part of this work has been performed using the PRISM core facility (Biogenouest©, UMS Biosit, Université de Rennes 35000 RENNES, FRANCE). Finally, Solenn Ferron is acknowledged for running 19F NMR experiments.
中文翻译:
酮上的氮杂王子环化获得具有四取代碳立构中心的哌啶
现代 Prins 环化涉及简单的起始原料,例如高烯丙醇或胺和醛,仅一步即可提供四氢吡喃或哌啶,同时形成碳-碳和碳-杂原子键。尽管 Prins 环化已得到充分记录,1 但其基于氮的环化却明显欠发达,2尽管哌啶环在天然产物和药物中比四氢吡喃更广泛。3尽管氮杂普林斯环化对于合成天然和/或生物活性化合物具有巨大潜力,但人们特别关注高烯丙胺的多样化。特别是,氮杂甲硅烷基 Prins 反应4已发展为在失去硅基团的情况下获得 3,4-脱氢哌啶,炔基氮杂-Prins 环化5提供了在 C-4 位具有亲核体的类似化合物,最后级联氮杂-据报道,Prins 反应6可形成稠合双环和三环系统。关于羰基伙伴,除了醛之外,迄今为止仅发表了两个例子:一个具有非常窄范围的酮7,另一个仅限于 1,2-二羰基底物8,两者都导致含有四取代基的哌啶的合成碳立构中心位于2位。
据我们所知,迄今为止尚未报道与非活化酮的有效氮杂王子环化。继续我们对 Prins 过程的兴趣,9我们在此公开了第一个与脂肪族、芳香族和杂环酮的氮杂-Prins 环化。值得注意的是,本方法与其他获得 2-螺哌啶的方法具有很好的竞争性。10
我们已经报道了路易斯酸和布朗斯台德酸的协同组合在仲烷基高烯丙胺和醛之间的氮杂普林斯环化中的有效性。11因此,我们首先研究了N-苄基高烯丙胺1和环己酮 ( 2a ) 在 1 当量存在下的反应性。TiCl 4和0.1当量。p -TsOH ⋅ H 2 O 在 DCM 中于 60 °C 在密封 Schlenk 管中培养 24 小时(表 1,条目 1)。不幸的是,令人失望的是缺乏反应性。接下来,我们研究了单独的 Brønsted 酸和 Lewis 酸的影响(表 1,分别为条目 2 和 3),在 1 当量存在下,仅观察到所需 2-螺哌啶 3a的转化率为 9%。通过粗反应混合物的1 H-NMR分析来确定TiCl 4的含量。受到这一结果的鼓舞,我们决定探索不同添加剂的效果,例如分子筛、水以及 PPh 3(表 1,条目 4-6),而无需进一步改进。令人欣慰的是,先使用 1 个当量,然后再使用 2 个当量。三乙胺 (TEA)(表 1,条目 7 和 8)分别导致 25% 和 52% 转化为所需产物。最后的结果促使我们研究二胺,特别是N , N , N' , N' -四甲基乙二胺 (TMEDA)、N , N , N' , N' -四乙基乙二胺 (TEEDA)、1,4-二氮杂双环2,2,2辛烷(DABCO) 和 1,2-二哌啶基乙烷。如表 1 中第 9-12 项所示,TEEDA 被发现更优越,导致 2-螺哌啶3a 的转化率为 61%,并被保留作为本研究其余部分的首选添加剂。为了进一步提高反应活性,接下来反应在 80 °C 的 1,2-DCE 中进行,粗混合物的分析确实显示 75% 的转化率(表 1,条目 13)。事实证明,再次将温度升高至100°C对反应不利,因为酮2a的自缩合/烯醇化产生的副产物变得过于丰富。最后,在反应过程中逐步添加过量的酮,即。e. 1.5当量 开始时然后是 0.5 当量。8 小时后,我们获得了90% 转化率的3 a(表 1,条目 14)。
|
||||
条目[一] |
添加剂(当量) |
溶剂 |
温度(°C) |
转化次数 (%) [b] |
|---|---|---|---|---|
1 |
p -TsOH·H 2 O (0.1) |
扩张型心肌病 |
60 |
0 |
2 [c] |
p -TsOH·H 2 O (1) |
扩张型心肌病 |
60 |
0 |
3 |
没有任何 |
扩张型心肌病 |
60 |
9 |
4 |
MS (3埃) |
扩张型心肌病 |
60 |
0 |
5 |
H 2 O (1) |
扩张型心肌病 |
60 |
0 |
6 |
PPh 3 (1) |
扩张型心肌病 |
60 |
0 |
7 |
茶 (1) |
扩张型心肌病 |
60 |
25 |
8 |
茶 (2) |
扩张型心肌病 |
60 |
52 |
9 |
TMEDA (1) |
扩张型心肌病 |
60 |
58 |
10 |
泰达 (1) |
扩张型心肌病 |
60 |
61 |
11 |
达伯科 (1) |
扩张型心肌病 |
60 |
43 |
12 |
1,2-二哌啶基 乙烷 (1) |
扩张型心肌病 |
60 |
55 |
13 |
泰达 (1) |
1,2-二氯乙烯 |
80 |
75 |
14 [d] |
泰达 (1) |
1,2-二氯乙烯 |
80 |
90 |
- [a]除非另有说明,反应均用高烯丙胺1(1 mmol,1 当量)、2 a(1 mmol,1 当量)和 TiCl 4(1 mmol,1 当量)在密封 Schlenk 管中进行24小时。[b]通过粗反应混合物的1 H-NMR分析计算转化率。[c]反应在没有TiCl 4 的情况下进行。[d]用1.5+0.5当量进行反应。2个。
在上述优化条件下,化合物3a 的分离率为 63%,首先评估了各种其他环酮的反应范围(方案 1)。环戊酮( 2b )和环庚酮( 2c )给出了令人满意的结果,尽管两个反应都需要在100℃下进行,并且产物3b和3c分别以30%和49%的产率分离。接下来,筛选了一系列在C-4位置2d - f取代的环己酮衍生物,无论该位置是否存在烷基或芳基,这些底物都很容易参与反应,生成3d、3e、和3f的产率分别为87%、57%和58%。必须强调的是,这一系列2-螺哌啶已观察到环烷异构现象,并且最稳定的反式异构体的形成是有利的,即使不是排他性的,如具有庞大的t -Bu基团的3e的情况。当存在时,两种异构体可通过硅胶柱色谱分离,并且3 f -反式的相对立体化学已通过单晶 X 射线分析明确证实,如图 1 所示。12
aza-Prins 环化的范围。反应条件:1 (1 mmol)、2 (1.5+0.5 当量)、TiCl 4 (1 当量)、TEEDA (1 当量) 于 1,2-DCE 中,在密封 Schlenk 管中于 80 °C 反应 24 小时。通过硅胶柱色谱纯化后测定产率(一般程序A)。[a]在 100 °C 下用2(1.5+1.5 当量)进行反应(一般程序B)。[b]没有 TEEDA 进行的反应(一般程序C)。
50% 热椭球体下3 f -反式(顶部)和3 l -反式(底部)的 ORTEP 表示。为了清楚起见,省略了氢原子。
然后将氮杂-普林斯环化应用于更具挑战性的无环底物。在 3-戊酮 ( 2 g )、4-庚酮 ( 2 h ) 和二苯甲酮 ( 2 i ) 存在下,我们很高兴地观察到化合物3 g – i的形成。由于其固有的构象迁移性,从无环系统构建四取代碳立构中心非常困难,13这里获得的产率(从 30% 到 34%)仍然是可以接受的。
受这些结果的鼓舞,我们接下来检查了N-苄基高烯丙胺1和各种活化的(二)酮之间的反应。我们满意地得出结论,该组有效地经历了氮杂-普林斯环化,具有非常显着的非对映选择性,有利于反式异构体。如方案1所示,苯偶酰( 2j )及其类似物4,4'-二甲基苯偶酰( 2k )得到可分离的非对映异构体3j和3k的混合物,总产率分别为64%和50%。使用酮酯2L可以以70%的产率回收3L-反式和3L-顺式,而三氟甲基化酮2M以60%的总产率得到哌啶3M-反式和3M-顺式。此外, 3 l -反式的单晶 X 射线分析可以确定反应过程中形成的两个立体中心的相对立体化学(图 1)。12
最后,我们研究了N-杂环酮的反应性。特别考虑哌啶酮,因为它们的反应可以提供螺稠合二哌啶的途径,这是一类具有强生物活性的化合物。14因此,在优化条件下尝试了1和 1-甲基-4-哌啶酮 ( 2 n )之间的 aza-Prins 环化,但导致反应活性损失。据推测,如果需要 TEEDA 来进行反应,则该添加剂和含氮基质的组合可能会使钛活性物质失活。事实上,在不向反应介质中添加 TEEDA 的情况下,反应活性得到恢复,并以 60% 的产率分离出产物3 n (方案 1)。这种趋势可以扩展到 1-benzyl-4-piperidone ( 2o ),导致相应的二哌啶3o,产率 49%。
总之,我们开发了一种钛-TEEDA 促进脂肪族、芳香族和杂环酮的氮杂-普林斯环化。我们的方法提供了在 C-2 位上带有四取代甚至螺环碳中心的哌啶的途径,这是常见的天然支架。当使用非对称底物和存在环烷异构体时,反应以非对映选择性进行,有利于反式异构体。我们的实验室正在进行进一步的应用和基材变化。
实验部分
一般程序A
向N-苄基高烯丙胺1 (163mL,1mmol)和酮(1.5mmol)在干燥1,2-DCE(10mL)中的溶液中添加TEEDA(213mL,1mmol)。将混合物搅拌10分钟并滴加TiCl 4溶液(1mL,1.0M在DCM中,1mmol)。将溶液在80℃搅拌8小时。冷却至室温后,再次将酮(0.5mmol)添加至混合物中并将溶液进一步在80℃搅拌过夜。冷却至室温后,用饱和NaHCO 3 淬灭反应。NaHCO 3 (15 mL)并在布氏漏斗(1层滤纸)上过滤。用DCM(2×20mL)萃取产物,用水(1×20mL)洗涤合并的有机相,经无水MgSO 4干燥,并蒸发至干燥。通过硅胶柱色谱法纯化残余物。
一般程序B
向N-苄基高烯丙胺1 (163mL,1mmol)和酮(1.5mmol)在干燥1,2-DCE(10mL)中的溶液中添加TEEDA(213mL,1mmol)。将混合物搅拌10分钟并滴加TiCl 4溶液(1mL,1.0M在DCM中,1mmol)。将溶液在100℃搅拌8小时。冷却至室温后,再次将酮(1.5mmol)添加至混合物中并将溶液进一步在100℃搅拌过夜。冷却至室温后,用饱和NaHCO 3 淬灭反应。NaHCO 3 (15 mL)并在布氏漏斗(1层滤纸)上过滤。用DCM(2×20mL)萃取产物,用水(1×20mL)洗涤合并的有机相,经无水MgSO 4干燥,并蒸发至干燥。通过硅胶柱色谱法纯化残余物。
一般程序C
将N-苄基高烯丙胺1 (163 mL, 1 mmol) 和酮 (1.5 mmol) 在干燥 1,2-DCE (10 mL) 中的溶液搅拌 10 分钟,然后添加 TiCl 4溶液 ( 1 mL, 1.0 M 的 DCM 溶液,1 mmol) 滴加。将溶液在80℃搅拌8小时。冷却至室温后,再次将酮(0.5mmol)添加至混合物中并将溶液进一步在80℃搅拌过夜。冷却至室温后,用饱和NaHCO 3 淬灭反应。NaHCO 3 (15 mL)并在布氏漏斗(1层滤纸)上过滤。用DCM(2×20mL)萃取产物,用水(1×20mL)洗涤合并的有机相,经无水MgSO 4干燥,并蒸发至干燥。通过硅胶柱色谱法纯化残余物。
致谢
作者感谢雷恩大学和法国国家科学研究中心的财政支持。衷心感谢雷恩化学研究所用于质量测量的 CRMPO、用于 X 射线分析的 Diffractométrie X 中心以及用于购买单晶设备的 FEDER 资金。这项工作的一部分是使用 PRISM 核心设施(Biogenouest©, UMS Biosit, Université de Rennes 35000 RENNES, FRANCE)进行的。最后,Solenn Ferron 因进行19 F NMR 实验而受到认可。
































 京公网安备 11010802027423号
京公网安备 11010802027423号