Computational and Theoretical Chemistry ( IF 3.0 ) Pub Date : 2023-11-18 , DOI: 10.1016/j.comptc.2023.114401 S. Selvakumari , Shine Kadaikunnan , Ghulam Abbas , S. Muthu
|
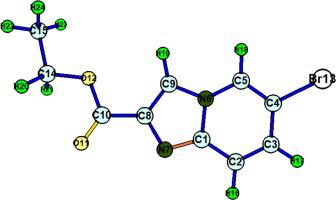
The current study emphasis on the structural characterization, solvation effects on electronic, thermodynamical properties and biological studies of Ethyl 6-bromoimidazo[1,2-a]pyridine-2-carboxylate (E6BP2C) employing DFT technique. Utilising 6–311++G(d,p) source set in DFT/B3LYP method, optimization of the structure & geometrical parameters were acquired. The resulting augmented geometrical factors are then utilized with an appropriate scaling factor to scale the probable vibrational wavenumbers. The MEP map disclosed the locations of electrophilic & nucleophilic regions. To describe the chemical reactivity, FMOs and Fukui function assessments have both been used. Multiwave function has been utilized to investigate topological analysis (ELF, LOL & RDG) and electron - hole dispersal for the excited states has also been pondered. The absorption of maximum wavelengths has been estimated with a variety of solvents utilizing the TD-DFT method and a virtual UV–Visible spectrum. Additionally, the chemical's optical and stability properties were ascertained using computational NBO and NLO investigations. 1.8765E-30 and 1.2688E-29 correspondingly are the 1st order hyperpolarizability values for gas and water phases. Thermodynamic parameters have also been calculated for variable solvents. ADMET along with drug likeness were used to determine the biologic characteristics.
中文翻译:
6-溴咪唑并[1,2-a]吡啶-2-甲酸乙酯的溶剂中电子激发性质、分子相互作用能、拓扑和生物学性质的计算研究
目前的研究重点是采用DFT技术对6-溴咪唑并[1,2-a]吡啶-2-甲酸乙酯(E6BP2C)进行结构表征、溶剂化对电子、热力学性质的影响以及生物学研究。利用DFT/B3LYP方法中的6–311++G(d,p)源集,获得了结构和几何参数的优化。然后将所得的增强几何因子与适当的缩放因子一起使用来缩放可能的振动波数。MEP 图揭示了亲电子和亲核区域的位置。为了描述化学反应性,FMO 和 Fukui 功能评估都被使用。多波函数已被用来研究拓扑分析(ELF、LOL 和 RDG),并且还考虑了激发态的电子-空穴扩散。利用 TD-DFT 方法和虚拟紫外-可见光谱估算了各种溶剂的最大波长吸收。此外,还通过计算 NBO 和 NLO 研究确定了该化学品的光学和稳定性特性。1.8765E-30 和 1.2688E-29 相应地是气相和水相的一阶超极化率值。还计算了可变溶剂的热力学参数。ADMET 与药物相似性一起用于确定生物学特性。






























 京公网安备 11010802027423号
京公网安备 11010802027423号