当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Enabling Nucleophilic Reactivity in High-Spin Fe(II) Imido Complexes: From Elementary Steps to Cooperative Catalysis
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2023-11-13 , DOI: 10.1021/acs.accounts.3c00511 Yafei Gao 1 , Jeremy M Smith 1
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2023-11-13 , DOI: 10.1021/acs.accounts.3c00511 Yafei Gao 1 , Jeremy M Smith 1
Affiliation
|
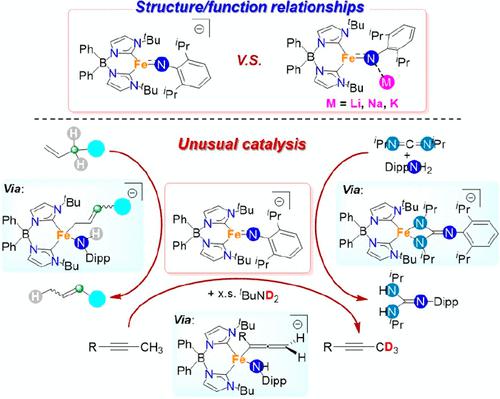
Transition metal complexes featuring an M═NR bond have received great attention as critical intermediates in the synthesis of nitrogen-containing compounds. In general, the properties of the imido ligand in these complexes are dependent on the nature of the metal center. Thus, the imido ligand tends to be nucleophilic in early transition metal complexes and electrophilic in late transition metal complexes. Nonetheless, the supporting ligand can have a dramatic effect on its reactivity. For example, there are sporadic examples of nucleophilic late transition metal imido complexes, often based on strongly donating supporting ligands. Building on these earlier works, in this Article, we show that the imido ligand in a low-coordinate high-spin bis(carbene)borate Fe(II) complex is able to access previously unknown reaction pathways, ultimately leading to new catalytic transformations. We first focus on the synthesis, characterization, and stoichiometric reactivity of a highly nucleophilic Fe(II) imido complex. The entry point for this system is the intermediate-spin three-coordinate Fe(III) imido complex, which is generated from the reaction of an Fe(I) synthon with an organic azide. Alkali metal reduction leads to a series of M+ (M = Li, Na, K) coordinated and charge-separated (M = K(18-C-6)) high-spin Fe(II) imido complexes, all of which have been isolated and fully characterized. Combined with the electronic structure calculations, these results reveal that the alkali ions moderately polarize the Fe═N bond according to K+ ≈ Na+ < Li+. As a result, the basicity of the imido ligand increases from the charged separated complex to K+, Na+, and Li+ coordinated complexes, as validated by intermolecular proton transfer equilibria. The impact of the counterion on imido ligand reactivity is demonstrated through protonation, alkylation, and hydrogen atom abstraction reactions. The counterion also directs the outcome of [2 + 2] reactions with benzophenone, where alkali coordination facilitates double bond metathesis. Building from here, we describe how the unusual nucleophilicity of the high-spin Fe(II) imido complex revealed in stoichiometric reactions can be extended to new catalytic transformations. For example, a [2 + 2] cycloaddition reaction serves as the basis for the catalytic guanylation of carbodiimides under mild conditions. More interestingly, this complex also exhibits the first ene-like reactivity of an M═NR bond in reactions with alkynes, nitriles, and alkenes. These transformations form the basis of catalytic alkyne and nitrile α-deuteration and pKa-dictated alkene transposition reactions, respectively. Mechanistic studies reveal the critical role of metal–ligand cooperativity in facilitating these catalytic transformations and suggest the new avenues for transition metal imido complexes in catalysis that extend beyond classical nitrene transfer chemistry.
中文翻译:

在高自旋 Fe(II) 亚氨基配合物中实现亲核反应:从基本步骤到协同催化
具有M=NR键的过渡金属配合物作为含氮化合物合成中的关键中间体受到了极大的关注。一般来说,这些配合物中亚氨基配体的性质取决于金属中心的性质。因此,亚氨基配体在早期过渡金属配合物中往往是亲核的,而在晚期过渡金属配合物中往往是亲电子的。尽管如此,支持配体可以对其反应性产生巨大影响。例如,有一些零星的亲核后过渡金属酰亚胺配合物的例子,通常基于强供体支持配体。在这些早期工作的基础上,在本文中,我们表明低配位高自旋双(碳烯)硼酸盐 Fe(II) 配合物中的酰亚胺配体能够进入以前未知的反应途径,最终导致新的催化转化。我们首先关注高亲核 Fe(II) 亚氨基配合物的合成、表征和化学计量反应性。该系统的入口点是中间自旋三配位 Fe(III) 亚氨基配合物,它是由 Fe(I) 合成子与有机叠氮化物反应生成的。碱金属还原产生一系列 M + (M = Li, Na, K) 配位和电荷分离 (M = K(18-C-6)) 高自旋 Fe(II) 亚氨基配合物,所有这些配合物都具有已被分离并充分表征。结合电子结构计算,这些结果表明碱离子根据 K + ≈ Na + < Li +适度极化 Fe=N 键。 结果,亚氨基配体的碱性从带电分离的络合物增加到 K + 、Na +和 Li +配位络合物,如分子间质子转移平衡所验证的。抗衡离子对亚氨基配体反应性的影响通过质子化、烷基化和氢原子夺取反应来证明。抗衡离子还指导与二苯甲酮的 [2 + 2] 反应的结果,其中碱配位促进双键复分解。在此基础上,我们描述了化学计量反应中揭示的高自旋 Fe(II) 亚氨基配合物的不寻常亲核性如何扩展到新的催化转化。例如,[2 + 2]环加成反应是在温和条件下碳二亚胺催化鸟苷化的基础。更有趣的是,该配合物在与炔烃、腈和烯烃的反应中还首次表现出M=NR键的类烯反应活性。这些转化分别形成催化炔烃和腈α-氘化以及p K a指示的烯烃转位反应的基础。机理研究揭示了金属-配体协同性在促进这些催化转化中的关键作用,并提出了过渡金属酰亚胺配合物在催化中超越经典氮宾转移化学的新途径。
更新日期:2023-11-13
中文翻译:
在高自旋 Fe(II) 亚氨基配合物中实现亲核反应:从基本步骤到协同催化
具有M=NR键的过渡金属配合物作为含氮化合物合成中的关键中间体受到了极大的关注。一般来说,这些配合物中亚氨基配体的性质取决于金属中心的性质。因此,亚氨基配体在早期过渡金属配合物中往往是亲核的,而在晚期过渡金属配合物中往往是亲电子的。尽管如此,支持配体可以对其反应性产生巨大影响。例如,有一些零星的亲核后过渡金属酰亚胺配合物的例子,通常基于强供体支持配体。在这些早期工作的基础上,在本文中,我们表明低配位高自旋双(碳烯)硼酸盐 Fe(II) 配合物中的酰亚胺配体能够进入以前未知的反应途径,最终导致新的催化转化。我们首先关注高亲核 Fe(II) 亚氨基配合物的合成、表征和化学计量反应性。该系统的入口点是中间自旋三配位 Fe(III) 亚氨基配合物,它是由 Fe(I) 合成子与有机叠氮化物反应生成的。碱金属还原产生一系列 M + (M = Li, Na, K) 配位和电荷分离 (M = K(18-C-6)) 高自旋 Fe(II) 亚氨基配合物,所有这些配合物都具有已被分离并充分表征。结合电子结构计算,这些结果表明碱离子根据 K + ≈ Na + < Li +适度极化 Fe=N 键。 结果,亚氨基配体的碱性从带电分离的络合物增加到 K + 、Na +和 Li +配位络合物,如分子间质子转移平衡所验证的。抗衡离子对亚氨基配体反应性的影响通过质子化、烷基化和氢原子夺取反应来证明。抗衡离子还指导与二苯甲酮的 [2 + 2] 反应的结果,其中碱配位促进双键复分解。在此基础上,我们描述了化学计量反应中揭示的高自旋 Fe(II) 亚氨基配合物的不寻常亲核性如何扩展到新的催化转化。例如,[2 + 2]环加成反应是在温和条件下碳二亚胺催化鸟苷化的基础。更有趣的是,该配合物在与炔烃、腈和烯烃的反应中还首次表现出M=NR键的类烯反应活性。这些转化分别形成催化炔烃和腈α-氘化以及p K a指示的烯烃转位反应的基础。机理研究揭示了金属-配体协同性在促进这些催化转化中的关键作用,并提出了过渡金属酰亚胺配合物在催化中超越经典氮宾转移化学的新途径。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号