当前位置:
X-MOL 学术
›
J. Mol. Struct.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Green synthesis, spectroscopic investigation, quantum chemical and molecular docking studies of 3-methylisoxazolo [4,5-b]pyridine
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2023-11-07 , DOI: 10.1016/j.molstruc.2023.136964 E. Dhanalakshmi , P. Rajesh , S. Suresh , M. Priyadharshini , M. Prabhaharan
Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2023-11-07 , DOI: 10.1016/j.molstruc.2023.136964 E. Dhanalakshmi , P. Rajesh , S. Suresh , M. Priyadharshini , M. Prabhaharan
|
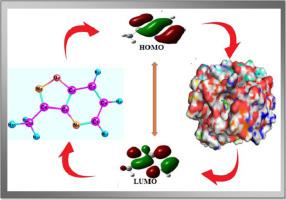
The present work, synthesized molecular compound of MIP from carbinol extract of by green synthesis method. The GC–MS analysis method has validated the molecular structure of MIP and revealed structural characteristics. FT-IR and UV-Visible spectra have been used to characterize the newly synthesized molecular molecule. DFT calculations employing the B3LYP/6-311++G(d,p) basis set are used to analyses the optimized molecular geometry and vibrational frequencies of the MIP compound. The Homo-Lumo energy gap of the MIP molecules is 5.4793 eV. The interaction (intra-inter, hydrogen bonding, and charge delocalization) of MIP molecules computed by NBO 3.1 program. TD-DFT was used to describe the electronic transition of ground state to excited state and compare with the recorded UV- Visible absorption spectra of MIP compounds. Molecular docking studies was interpreted and recorded the newly synthesis compound against 2YJR and IUSN receptor (lungs and kidney cancer).
中文翻译:

3-甲基异恶唑并[4,5-b]吡啶的绿色合成、光谱研究、量子化学和分子对接研究
本工作采用绿色合成方法从甲醇提取物中合成了MIP分子化合物。 GC-MS分析方法验证了MIP的分子结构并揭示了结构特征。 FT-IR 和 UV-Visible 光谱已用于表征新合成的分子。采用 B3LYP/6-311++G(d,p) 基组的 DFT 计算用于分析 MIP 化合物的优化分子几何形状和振动频率。 MIP 分子的 Homo-Lumo 能隙为 5.4793 eV。通过 NBO 3.1 程序计算 MIP 分子的相互作用(间内、氢键和电荷离域)。 TD-DFT 用于描述基态到激发态的电子跃迁,并与记录的 MIP 化合物的紫外-可见吸收光谱进行比较。分子对接研究解释并记录了针对 2YJR 和 IUSN 受体(肺癌和肾癌)的新合成化合物。
更新日期:2023-11-07
中文翻译:
3-甲基异恶唑并[4,5-b]吡啶的绿色合成、光谱研究、量子化学和分子对接研究
本工作采用绿色合成方法从甲醇提取物中合成了MIP分子化合物。 GC-MS分析方法验证了MIP的分子结构并揭示了结构特征。 FT-IR 和 UV-Visible 光谱已用于表征新合成的分子。采用 B3LYP/6-311++G(d,p) 基组的 DFT 计算用于分析 MIP 化合物的优化分子几何形状和振动频率。 MIP 分子的 Homo-Lumo 能隙为 5.4793 eV。通过 NBO 3.1 程序计算 MIP 分子的相互作用(间内、氢键和电荷离域)。 TD-DFT 用于描述基态到激发态的电子跃迁,并与记录的 MIP 化合物的紫外-可见吸收光谱进行比较。分子对接研究解释并记录了针对 2YJR 和 IUSN 受体(肺癌和肾癌)的新合成化合物。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号