Apoptosis ( IF 6.1 ) Pub Date : 2023-11-09 , DOI: 10.1007/s10495-023-01911-8
Daria Monogiou Belik 1 , Riccardo Bernasconi 1 , Lifen Xu 1 , Giacomo Della Verde 1 , Vera Lorenz 1 , Vivienne Grüterich 1 , Melania Balzarolo 1 , Michika Mochizuki 1 , Otmar Pfister 1, 2 , Gabriela M Kuster 1, 2
|
Background
Tyrosine kinase inhibitors (TKIs) targeting fms-like tyrosine kinase 3 (Flt3) such as quizartinib were specifically designed for acute myeloid leukemia treatment, but also multi-targeting TKIs applied to solid tumor patients inhibit Flt3. Flt3 is expressed in the heart and its activation is cytoprotective in myocardial infarction (MI) in mice.
Objectives
We sought to test whether Flt3-targeting TKI treatment aggravates cardiac injury after MI.
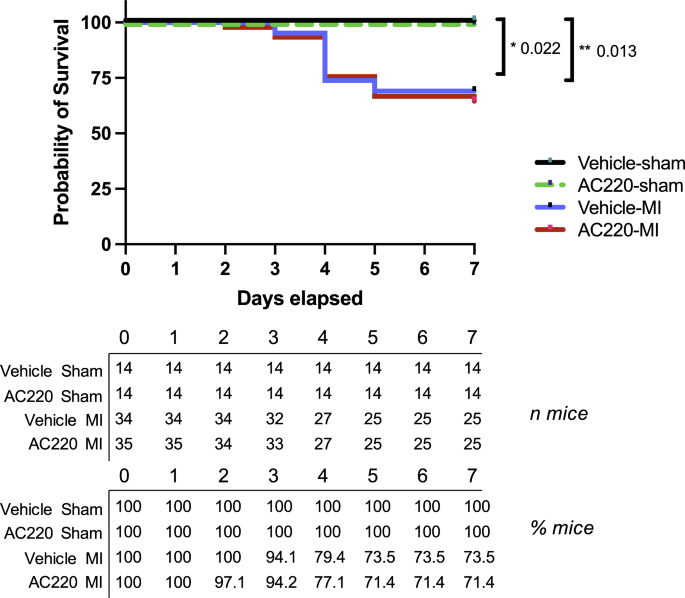
Methods and results
Compared to vehicle, quizartinib (10 mg/kg/day, gavage) did not alter cardiac dimensions or function in healthy mice after four weeks of therapy. Pretreated mice were randomly assigned to MI or sham surgery while receiving quizartinib or vehicle for one more week. Quizartinib did not aggravate the decline in ejection fraction, but significantly enhanced ventricular dilatation one week after infarction. In addition, apoptotic cell death was significantly increased in the myocardium of quizartinib-treated compared to vehicle-treated mice. In vitro, quizartinib dose-dependently decreased cell viability in neonatal rat ventricular myocytes and in H9c2 cells, and increased apoptosis as assessed in the latter. Together with H2O2, quizartinib potentiated the phosphorylation of the pro-apoptotic mitogen activated protein kinase p38 and augmented H2O2-induced cell death and apoptosis beyond additive degree. Pretreatment with a p38 inhibitor abolished apoptosis under quizartinib and H2O2.
Conclusion
Quizartinib potentiates apoptosis and promotes maladaptive remodeling after MI in mice at least in part via a p38-dependent mechanism. These findings are consistent with the multi-hit hypothesis of cardiotoxicity and make cardiac monitoring in patients with ischemic heart disease under Flt3- or multi-targeting TKIs advisable.
中文翻译:
Flt3 抑制剂 quizartinib 可增强小鼠心肌梗死后细胞凋亡并促进适应不良重塑
背景
靶向 fms 样酪氨酸激酶 3 (Flt3) 的酪氨酸激酶抑制剂 (TKI)(例如 quizartinib)专为急性髓性白血病治疗而设计,但应用于实体瘤患者的多靶点 TKI 也能抑制 Flt3。 Flt3 在心脏中表达,其激活对小鼠心肌梗塞 (MI) 具有细胞保护作用。
目标
我们试图测试 Flt3 靶向 TKI 治疗是否会加重 MI 后的心脏损伤。
方法和结果
与载体相比,治疗 4 周后,quizartinib(10 mg/kg/天,强饲法)并未改变健康小鼠的心脏尺寸或功能。预处理的小鼠被随机分配接受 MI 或假手术,同时接受 quizartinib 或媒介物治疗一周。 Quizartinib并没有加重射血分数的下降,但在梗塞后1周显着增强心室扩张。此外,与媒介物治疗的小鼠相比,奎扎替尼治疗的小鼠心肌中的凋亡细胞死亡显着增加。在体外,quizartinib 剂量依赖性地降低新生大鼠心室肌细胞和 H9c2 细胞的细胞活力,并增加后者的细胞凋亡。 quizartinib 与 H 2 O 2一起增强促凋亡丝裂原激活蛋白激酶 p38 的磷酸化,并以累加程度增强 H 2 O 2诱导的细胞死亡和凋亡。 p38 抑制剂预处理在 quizartinib 和 H 2 O 2作用下消除了细胞凋亡。
结论
Quizartinib 至少部分通过 p38 依赖性机制增强细胞凋亡并促进 MI 后小鼠的适应不良重塑。这些发现与心脏毒性的多重打击假说一致,并建议在 Flt3 或多靶点 TKI 治疗下对缺血性心脏病患者进行心脏监测。
































 京公网安备 11010802027423号
京公网安备 11010802027423号