当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate and Efficient Spin–Phonon Coupling and Spin Dynamics Calculations for Molecular Solids
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-11-02 , DOI: 10.1021/jacs.3c06015 Rizwan Nabi 1 , Jakob K Staab 1 , Andrea Mattioni 1 , Jon G C Kragskow 1, 2 , Daniel Reta 1, 3, 4, 5 , Jonathan M Skelton 1 , Nicholas F Chilton 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-11-02 , DOI: 10.1021/jacs.3c06015 Rizwan Nabi 1 , Jakob K Staab 1 , Andrea Mattioni 1 , Jon G C Kragskow 1, 2 , Daniel Reta 1, 3, 4, 5 , Jonathan M Skelton 1 , Nicholas F Chilton 1
Affiliation
|
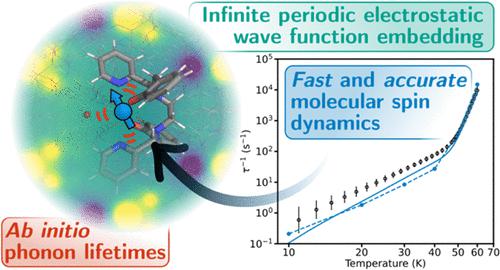
Molecular materials are poised to play a significant role in the development of future optoelectronic and quantum technologies. A crucial aspect of these areas is the role of spin–phonon coupling and how it facilitates energy transfer processes such as intersystem crossing, quantum decoherence, and magnetic relaxation. Thus, it is of significant interest to be able to accurately calculate the molecular spin–phonon coupling and spin dynamics in the condensed phase. Here, we demonstrate the maturity of ab initio methods for calculating spin–phonon coupling by performing a case study on a single-molecule magnet and showing quantitative agreement with the experiment, allowing us to explore the underlying origins of its spin dynamics. This feat is achieved by leveraging our recent developments in analytic spin–phonon coupling calculations in conjunction with a new method for including the infinite electrostatic potential in the calculations. Furthermore, we make the first ab initio determination of phonon lifetimes and line widths for a molecular magnet to prove that the commonplace Born–Markov assumption for the spin dynamics is valid, but such “exact” phonon line widths are not essential to obtain accurate magnetic relaxation rates. Calculations using this approach are facilitated by the open-source packages we have developed, enabling cost-effective and accurate spin–phonon coupling calculations on molecular solids.
中文翻译:

分子固体的准确高效的自旋声子耦合和自旋动力学计算
分子材料有望在未来光电和量子技术的发展中发挥重要作用。这些领域的一个重要方面是自旋声子耦合的作用以及它如何促进能量传递过程,例如系间跨越、量子退相干和磁弛豫。因此,能够准确计算凝聚相中的分子自旋声子耦合和自旋动力学具有重要意义。在这里,我们通过对单分子磁体进行案例研究并显示与实验的定量一致性,展示了计算自旋声子耦合的从头计算方法的成熟度,使我们能够探索其自旋动力学的潜在起源。这一壮举是通过利用我们在分析自旋声子耦合计算方面的最新进展以及在计算中包含无限静电势的新方法来实现的。此外,我们首次从头算确定了分子磁体的声子寿命和线宽,以证明自旋动力学的常见玻恩-马尔可夫假设是有效的,但这种“精确”的声子线宽对于获得精确的磁力并不是必需的。松弛率。我们开发的开源软件包促进了使用这种方法的计算,从而能够对分子固体进行经济有效且准确的自旋声子耦合计算。
更新日期:2023-11-02
中文翻译:
分子固体的准确高效的自旋声子耦合和自旋动力学计算
分子材料有望在未来光电和量子技术的发展中发挥重要作用。这些领域的一个重要方面是自旋声子耦合的作用以及它如何促进能量传递过程,例如系间跨越、量子退相干和磁弛豫。因此,能够准确计算凝聚相中的分子自旋声子耦合和自旋动力学具有重要意义。在这里,我们通过对单分子磁体进行案例研究并显示与实验的定量一致性,展示了计算自旋声子耦合的从头计算方法的成熟度,使我们能够探索其自旋动力学的潜在起源。这一壮举是通过利用我们在分析自旋声子耦合计算方面的最新进展以及在计算中包含无限静电势的新方法来实现的。此外,我们首次从头算确定了分子磁体的声子寿命和线宽,以证明自旋动力学的常见玻恩-马尔可夫假设是有效的,但这种“精确”的声子线宽对于获得精确的磁力并不是必需的。松弛率。我们开发的开源软件包促进了使用这种方法的计算,从而能够对分子固体进行经济有效且准确的自旋声子耦合计算。
































 京公网安备 11010802027423号
京公网安备 11010802027423号