当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Excited-state relaxation mechanisms of 2,2′-(1-phenyl-1H-1,2,4-triazole-3,5-diyl)diphenol: single- or double-proton transfer?
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2023-10-26 , DOI: 10.1039/d3cp02680a Jiahui Wu 1 , Jihuan He 1 , Wei Wang 1 , Xiaohang Chen 1 , Shu-Hua Xia 1
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2023-10-26 , DOI: 10.1039/d3cp02680a Jiahui Wu 1 , Jihuan He 1 , Wei Wang 1 , Xiaohang Chen 1 , Shu-Hua Xia 1
Affiliation
|
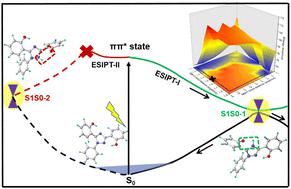
Triazole compounds are important organic systems with excellent electronic properties, which have diagnostic potential in the fields of organic electronics and organic photovoltaics. The important photophysical nature of these systems is the transformation between the enol and keto forms after excited-state proton transfer. In this study, the IR vibrational spectrum, ESIPT mechanism, and excited-state decay dynamics of 2,2′-(1-phenyl-1H-1,2,4-triazole-3,5-diyl)diphenol (ExPh) were explored using electronic structure calculations and non-adiabatic dynamics simulations. Two S1/S0 conical intersections with distinct proton transfer (ESIPT-I and ESIPT-II) involved were obtained. The associated two-dimensional S1 minimum-energy potential energy surface indicated that the dynamical roles of these two S1/S0 conical intersections in the S1 excited-state decay were quite different. The ESIPT-I reaction was more favorable to occur than the ESIPT-II process. Our dynamics simulations supported this hypothesis with the whole trajectories decaying to the ground state via the S1S0-1 conical intersection, which involved the ESIPT-I process. The ESIPT-Involved efficient deactivation pathway could be partially responsible for the decrease in fluorescence emission. These results and ESIPT mechanisms are helpful for understanding the decay pathways of similar systems.
中文翻译:

2,2′-(1-苯基-1H-1,2,4-三唑-3,5-二基)联苯酚的激发态弛豫机制:单质子转移还是双质子转移?
三唑类化合物是重要的有机体系,具有优异的电子性能,在有机电子和有机光伏领域具有诊断潜力。这些系统的重要光物理性质是激发态质子转移后烯醇和酮形式之间的转变。在本研究中,2,2′-(1-苯基-1 H -1,2,4-三唑-3,5-二基)联苯酚 (ExPh) 的红外振动光谱、ESIPT 机制和激发态衰变动力学使用电子结构计算和非绝热动力学模拟来探索。获得了两个涉及不同质子转移的S 1 /S 0圆锥形交叉点( ESIPT-I和ESIPT-II )。相关的二维S 1最小能量势能面表明这两个S 1 /S 0圆锥形交叉点在S 1激发态衰变中的动力学作用有很大不同。 ESIPT-I反应比ESIPT-II过程更有利于发生。我们的动力学模拟支持了这一假设,整个轨迹通过S1S0-1 圆锥形交叉点衰减到基态,这涉及ESIPT-I过程。 ESIPT涉及的有效失活途径可能是荧光发射减少的部分原因。这些结果和 ESIPT 机制有助于理解类似系统的衰变路径。
更新日期:2023-10-26
中文翻译:
2,2′-(1-苯基-1H-1,2,4-三唑-3,5-二基)联苯酚的激发态弛豫机制:单质子转移还是双质子转移?
三唑类化合物是重要的有机体系,具有优异的电子性能,在有机电子和有机光伏领域具有诊断潜力。这些系统的重要光物理性质是激发态质子转移后烯醇和酮形式之间的转变。在本研究中,2,2′-(1-苯基-1 H -1,2,4-三唑-3,5-二基)联苯酚 (ExPh) 的红外振动光谱、ESIPT 机制和激发态衰变动力学使用电子结构计算和非绝热动力学模拟来探索。获得了两个涉及不同质子转移的S 1 /S 0圆锥形交叉点( ESIPT-I和ESIPT-II )。相关的二维S 1最小能量势能面表明这两个S 1 /S 0圆锥形交叉点在S 1激发态衰变中的动力学作用有很大不同。 ESIPT-I反应比ESIPT-II过程更有利于发生。我们的动力学模拟支持了这一假设,整个轨迹通过S1S0-1 圆锥形交叉点衰减到基态,这涉及ESIPT-I过程。 ESIPT涉及的有效失活途径可能是荧光发射减少的部分原因。这些结果和 ESIPT 机制有助于理解类似系统的衰变路径。
































 京公网安备 11010802027423号
京公网安备 11010802027423号