当前位置:
X-MOL 学术
›
J. Am. Soc. Mass Spectrom.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An Optimized and High-Throughput Method for Histone Propionylation and Data-Independent Acquisition Analysis for the Identification and Quantification of Histone Post-translational Modifications
Journal of the American Society for Mass Spectrometry ( IF 3.1 ) Pub Date : 2023-10-18 , DOI: 10.1021/jasms.3c00223
Richard M Searfoss 1 , Rashmi Karki 1 , Zongtao Lin 1 , Faith Robison 1 , Benjamin A Garcia 1
Journal of the American Society for Mass Spectrometry ( IF 3.1 ) Pub Date : 2023-10-18 , DOI: 10.1021/jasms.3c00223
Richard M Searfoss 1 , Rashmi Karki 1 , Zongtao Lin 1 , Faith Robison 1 , Benjamin A Garcia 1
Affiliation
|
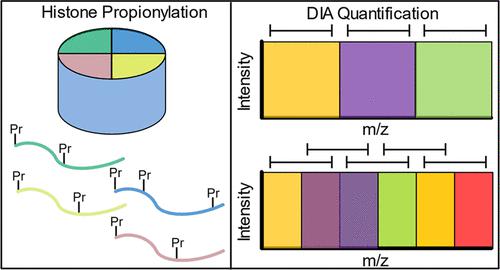
Histones are DNA binding proteins that allow for packaging of the DNA into the nucleus. They are abundantly present across the genome and thus serve as a major site of epigenetic regulation through the use of post-translational modifications (PTMs). Aberrations in histone expression and modifications have been implicated in a variety of human diseases and thus are a major focus of disease etiology studies. A well-established method for studying histones and PTMs is through the chemical derivatization of isolated histones followed by liquid chromatography and mass spectrometry analysis. Using such an approach has allowed for a swath of discoveries to be found, leading to novel therapeutics such as histone deacetylase (HDAC) inhibitors that have already been applied in the clinic. However, with the rapid improvement in instrumentation and data analysis pipelines, it remains important to temporally re-evaluate the established protocols to improve throughput and ensure data quality. Here, we optimized the histone derivatization procedure to increase sample throughput without compromising peptide quantification. An implemented spike-in standard peptide further serves as a quality control to evaluate the propionylation and digestion efficiencies as well as reproducibility in chromatographic retention and separation. Last, the application of various data-independent acquisition (DIA) strategies was explored to ensure low variation between runs. The output of this study is a newly optimized derivatization protocol and mass spectrometry method that maintains high identification and quantification of histone PTMs while increasing sample throughput.
中文翻译:

一种优化的高通量组蛋白丙酰化方法和数据独立的采集分析,用于组蛋白翻译后修饰的鉴定和定量
组蛋白是 DNA 结合蛋白,可将 DNA 包装到细胞核中。它们大量存在于整个基因组中,因此通过使用翻译后修饰 (PTM) 作为表观遗传调控的主要位点。组蛋白表达和修饰的异常与多种人类疾病有关,因此是疾病病因学研究的主要焦点。研究组蛋白和 PTM 的一种行之有效的方法是对分离的组蛋白进行化学衍生化,然后进行液相色谱和质谱分析。使用这种方法已经取得了一系列的发现,从而催生了新的治疗方法,例如已经应用于临床的组蛋白脱乙酰酶 (HDAC) 抑制剂。然而,随着仪器和数据分析管道的快速改进,暂时重新评估已建立的协议以提高吞吐量并确保数据质量仍然很重要。在这里,我们优化了组蛋白衍生化程序,以在不影响肽定量的情况下提高样品通量。实施的掺入标准肽进一步用作质量控制,以评估丙酰化和消化效率以及色谱保留和分离的再现性。最后,探索了各种数据独立采集(DIA)策略的应用,以确保运行之间的低变化。本研究的成果是一种新优化的衍生化方案和质谱方法,可保持组蛋白 PTM 的高度识别和定量,同时提高样品通量。
更新日期:2023-10-18
中文翻译:
一种优化的高通量组蛋白丙酰化方法和数据独立的采集分析,用于组蛋白翻译后修饰的鉴定和定量
组蛋白是 DNA 结合蛋白,可将 DNA 包装到细胞核中。它们大量存在于整个基因组中,因此通过使用翻译后修饰 (PTM) 作为表观遗传调控的主要位点。组蛋白表达和修饰的异常与多种人类疾病有关,因此是疾病病因学研究的主要焦点。研究组蛋白和 PTM 的一种行之有效的方法是对分离的组蛋白进行化学衍生化,然后进行液相色谱和质谱分析。使用这种方法已经取得了一系列的发现,从而催生了新的治疗方法,例如已经应用于临床的组蛋白脱乙酰酶 (HDAC) 抑制剂。然而,随着仪器和数据分析管道的快速改进,暂时重新评估已建立的协议以提高吞吐量并确保数据质量仍然很重要。在这里,我们优化了组蛋白衍生化程序,以在不影响肽定量的情况下提高样品通量。实施的掺入标准肽进一步用作质量控制,以评估丙酰化和消化效率以及色谱保留和分离的再现性。最后,探索了各种数据独立采集(DIA)策略的应用,以确保运行之间的低变化。本研究的成果是一种新优化的衍生化方案和质谱方法,可保持组蛋白 PTM 的高度识别和定量,同时提高样品通量。
































 京公网安备 11010802027423号
京公网安备 11010802027423号