Current Treatment Options in Rheumatology Pub Date : 2023-10-18 , DOI: 10.1007/s40674-023-00213-z
Priyanka Verma , Swarna Bale , John Varga , Swati Bhattacharyya
|
Purpose of review
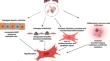
Systemic sclerosis–associated interstitial lung disease (SSc-ILD) and idiopathic pulmonary fibrosis (IPF) are fatal fibrotic lung disorders characterized by progressive fibrosis, immune dysregulation, vascular damage, fibroblast activation, and myofibroblast differentiation. In this review, we will briefly update all the above-mentioned mechanisms, cell-type involvement from single cell-RNA sequencing, and the differences with emerging common biomarkers and available treatment options.
Recent findings
In the IPF, tissue injury and lung fibrosis are thought to be initiated by injury of the alveolar epithelium and activation of alveolar epithelial cells (AECs) that release cytokines to activate the fibroblasts. By contrast, vascular damage is an early event in the pathogenesis of SSc-ILD and several diverse cells, such as myofibroblasts, fibroblasts, and endothelial cells, participate in inflammatory activation. Recent findings from single-cell RNA-seq suggested the presence of different subsets of innate and adaptive immune cells and differentiation of myofibroblasts from distinct sources, which complicates the understanding of the disease landscape. Fibroblast activation and myofibroblast accumulation are the final common pathways of lung fibrosis in both SSc-associated ILD and IPF.
Summary
Fibrosis in IPF appears to be related to aberrant repair following injury, but whether this also holds for SSc-ILD is less evident. Additionally, compared to IPF, immune dysregulation appears to contribute more to profibrotic responses in SSc-ILD. Nevertheless, persistent myofibroblast activity leads to progressive tissue fibrosis leading to organ failure and death. Few FDA-approved drugs (nintedanib, tocilizumab, and pirfenidone) are available for the treatment of fibrotic lung disease. Prospective high-throughput strategy-based approaches are required to understand the complexity of the diseases and develop specific therapeutic targets as part of precision medicine.
中文翻译:
系统性硬化症相关间质性肺病和特发性肺纤维化的分子机制:最新进展
审查目的
系统性硬化症相关间质性肺病(SSc-ILD)和特发性肺纤维化(IPF)是致命的纤维化性肺病,其特征是进行性纤维化、免疫失调、血管损伤、成纤维细胞活化和肌成纤维细胞分化。在这篇综述中,我们将简要更新所有上述机制、单细胞 RNA 测序涉及的细胞类型,以及与新兴常见生物标志物和可用治疗方案的差异。
最近的发现
在 IPF 中,组织损伤和肺纤维化被认为是由肺泡上皮损伤和肺泡上皮细胞 (AEC) 激活引发的,肺泡上皮细胞释放细胞因子以激活成纤维细胞。相比之下,血管损伤是 SSc-ILD 发病机制的早期事件,多种不同的细胞,如肌成纤维细胞、成纤维细胞和内皮细胞,参与炎症激活。单细胞 RNA 测序的最新发现表明,存在不同的先天性和适应性免疫细胞亚群,以及不同来源的肌成纤维细胞的分化,这使得对疾病状况的理解变得复杂。成纤维细胞活化和肌成纤维细胞积聚是 SSc 相关 ILD 和 IPF 肺纤维化的最终共同途径。
概括
IPF 中的纤维化似乎与损伤后的异常修复有关,但这是否也适用于 SSc-ILD 则不太明显。此外,与 IPF 相比,免疫失调似乎更能促进 SSc-ILD 的促纤维化反应。然而,持续的肌成纤维细胞活性会导致进行性组织纤维化,导致器官衰竭和死亡。 FDA 批准的药物(尼达尼布、托珠单抗和吡非尼酮)很少可用于治疗纤维化肺病。需要基于前瞻性高通量策略的方法来了解疾病的复杂性并开发特定的治疗靶点作为精准医学的一部分。
































 京公网安备 11010802027423号
京公网安备 11010802027423号