当前位置:
X-MOL 学术
›
J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
使用大规模计算机 ADMET 模型预测小分子可开发性
Journal of Medicinal Chemistry ( IF 6.8 ) Pub Date : 2023-10-10 , DOI: 10.1021/acs.jmedchem.3c01083
Maximilian Beckers 1 , Noé Sturm 1 , Finton Sirockin 1 , Nikolas Fechner 1 , Nikolaus Stiefl 1
Journal of Medicinal Chemistry ( IF 6.8 ) Pub Date : 2023-10-10 , DOI: 10.1021/acs.jmedchem.3c01083
Maximilian Beckers 1 , Noé Sturm 1 , Finton Sirockin 1 , Nikolas Fechner 1 , Nikolaus Stiefl 1
Affiliation
|
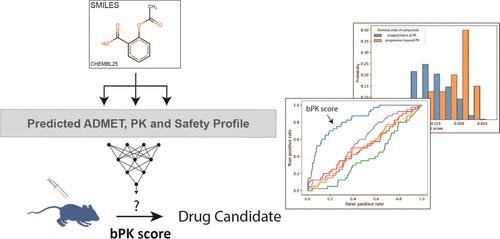
对一系列化合物输送药物的潜力进行早期计算机评估是计算机辅助药物设计的主要挑战之一。目标是从大的化学空间中识别出正确的化学系列化合物,然后优先考虑最有可能成为药物的分子。尽管几十年来已经开发出多种评估化合物的方法,但这些预测因子的质量往往不够好,并且与各自估计值一致的化合物不一定是药物样的。在这里,我们报告了一种新颖的深度学习方法,该方法利用~100 ADMET 检测的大规模预测来评估化合物成为相关候选药物的潜力。由此产生的分数,我们称之为 bPK 分数,大大优于以前的方法,并且在数据集上表现出很强的区分性能,而以前的方法则没有。

"点击查看英文标题和摘要"
更新日期:2023-10-10
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号