当前位置:
X-MOL 学术
›
Appl. Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical investigation of solvation and inter-chain interactions for phosphate ester dispersants on tetragonal BaTiO3 (0 0 1) in non-aqueous solvents: An ab initio molecular dynamics simulation approach
Applied Surface Science ( IF 6.3 ) Pub Date : 2023-10-10 , DOI: 10.1016/j.apsusc.2023.158645 Hee-Joon Chun , Inkyung Kim , Juhun Park , Giwoong Ha
Applied Surface Science ( IF 6.3 ) Pub Date : 2023-10-10 , DOI: 10.1016/j.apsusc.2023.158645 Hee-Joon Chun , Inkyung Kim , Juhun Park , Giwoong Ha
|
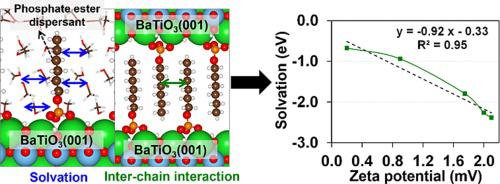
Tetragonal BaTiO is a key material for manufacturing multi-layer ceramic capacitors (MLCCs), and a highly dispersed BaTiO suspension, where the surfaces are conjugated by phosphate ester dispersants, is an essential technology for the production of ultra-thin films and small-sized ceramic capacitors. Therefore, understanding the interactions of conjugated polymers with solvents is critical to designing improved dispersants and solvents for commercial MLCC applications. In this work, we theoretically investigate the solvation and inter-chain interactions of mono-alkyl and ethoxy phosphate esters under methanol, ethanol, isopropanol, 1-butanol, and toluene, on tetragonal BaTiO (001) using the molecular dynamics (AIMD) simulations. Our results show that the total number of solvent molecules surrounding mono-alkyl and ethoxy phosphate esters decreases in the order of methanol > ethanol > isopropanol > 1-butanol > toluene, indicating that solvation via van der Waals forces and hydrogen bonding declines with increasing the hydrophobicity of solvent molecules. Also, the solvation interaction energy has a linear correlation with the experimental differential of zeta potential, suggesting that the calculated energy is associated with electrostatic environment at the dispersant/solvent/BaTiO interface. Therefore, the solvation interaction energy is an important factor that describes the liquid–solid interfaces as well as a dispersion stability of BaTiO suspension.
中文翻译:

非水溶剂中磷酸酯分散剂在四方 BaTiO3 (0 0 1) 上的溶剂化和链间相互作用的理论研究:从头算分子动力学模拟方法
四方钛酸钡是制造多层陶瓷电容器(MLCC)的关键材料,表面通过磷酸酯分散剂共轭的高度分散的钛酸钡悬浮液是生产超薄膜和小尺寸的关键技术。陶瓷电容器。因此,了解共轭聚合物与溶剂的相互作用对于为商业 MLCC 应用设计改进的分散剂和溶剂至关重要。在这项工作中,我们利用分子动力学 (AIMD) 模拟,从理论上研究了四方 BaTiO (001) 上单烷基和乙氧基磷酸酯在甲醇、乙醇、异丙醇、1-丁醇和甲苯下的溶剂化和链间相互作用。我们的结果表明,单烷基和乙氧基磷酸酯周围的溶剂分子总数按甲醇>乙醇>异丙醇>1-丁醇>甲苯的顺序减少,这表明通过范德华力和氢键的溶剂化作用随着溶剂分子的增加而减少。溶剂分子的疏水性。此外,溶剂化相互作用能与 zeta 电位的实验微分具有线性相关性,表明计算的能量与分散剂/溶剂/BaTiO 界面的静电环境有关。因此,溶剂化相互作用能是描述液固界面以及BaTiO悬浮液分散稳定性的重要因素。
更新日期:2023-10-10
中文翻译:
非水溶剂中磷酸酯分散剂在四方 BaTiO3 (0 0 1) 上的溶剂化和链间相互作用的理论研究:从头算分子动力学模拟方法
四方钛酸钡是制造多层陶瓷电容器(MLCC)的关键材料,表面通过磷酸酯分散剂共轭的高度分散的钛酸钡悬浮液是生产超薄膜和小尺寸的关键技术。陶瓷电容器。因此,了解共轭聚合物与溶剂的相互作用对于为商业 MLCC 应用设计改进的分散剂和溶剂至关重要。在这项工作中,我们利用分子动力学 (AIMD) 模拟,从理论上研究了四方 BaTiO (001) 上单烷基和乙氧基磷酸酯在甲醇、乙醇、异丙醇、1-丁醇和甲苯下的溶剂化和链间相互作用。我们的结果表明,单烷基和乙氧基磷酸酯周围的溶剂分子总数按甲醇>乙醇>异丙醇>1-丁醇>甲苯的顺序减少,这表明通过范德华力和氢键的溶剂化作用随着溶剂分子的增加而减少。溶剂分子的疏水性。此外,溶剂化相互作用能与 zeta 电位的实验微分具有线性相关性,表明计算的能量与分散剂/溶剂/BaTiO 界面的静电环境有关。因此,溶剂化相互作用能是描述液固界面以及BaTiO悬浮液分散稳定性的重要因素。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号