The Journal of Nutritional Biochemistry ( IF 4.8 ) Pub Date : 2023-09-23 , DOI: 10.1016/j.jnutbio.2023.109452
Yulong Gong 1 , Qisheng Lu 2 , Longwei Xi 2 , Yulong Liu 2 , Bingyuan Yang 3 , Jingzhi Su 2 , Haokun Liu 1 , Junyan Jin 1 , Zhimin Zhang 1 , Yunxia Yang 1 , Xiaoming Zhu 1 , Shouqi Xie 4 , Dong Han 5
|
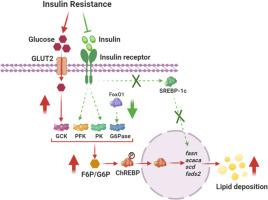
Insulin-sensitive lipogenesis dominates the body lipid deposition; however, nonalcoholic fatty liver disease (NAFLD) develops in the insulin-resistant state. The regulation mechanism of insulin resistance-driven NAFLD remains elusive. Using zebrafish model of insulin resistance (ZIR, insrb−/−) and mouse hepatocytes (NCTC 1469), we explored the regulation mechanism of insulin resistance-driven hepatic lipid deposition under the stimulation of carbohydrate diet (CHD). In ZIR model, insulin resistance induced hyperlipidemia and elevated hepatic lipid deposition via elevating the gene/protein expressions of lipogenic enzymes, that was activated by carbohydrate response element binding protein (ChREBP), rather than sterol regulatory element binding proteins 1c (SREBP-1c). The metabolomic analysis in zebrafish and silencing of chrebp in mouse hepatocytes revealed that the increased hepatic frucotose-6-phosphate (F6P) and glucose-6-phosphate (G6P) promoted the ChREBP-mediated lipid deposition. We further identified that F6P alone was sufficient to activate ChREBP-mediated lipid deposition by a SREBP-1c-independent manner. Moreover, we clarified the suppressed hepatic phosphofructokinase/glucose-6-phosphatase functions and the normal glucokinase function preserved by glucose transporter 2 (GLUT2) manipulated the increased F6P/G6P content in ZIR. In conclusion, the present study revealed that insulin resistance promoted hepatic lipid deposition via the F6P/G6P-mediated ChREBP activation. Our findings deciphered the main regulation pathway for the liver lipid deposition in the insulin-resistant state and identified F6P as a new potential regulator for ChREBP.
中文翻译:
F6P/G6P介导的ChREBP激活促进斑马鱼胰岛素抵抗驱动的肝脏脂质沉积
胰岛素敏感性脂肪生成主导体内脂质沉积;然而,非酒精性脂肪肝(NAFLD)是在胰岛素抵抗状态下发生的。胰岛素抵抗驱动的 NAFLD 的调节机制仍不清楚。利用斑马鱼胰岛素抵抗模型(ZIR, insrb −/−)和小鼠肝细胞(NCTC 1469),我们探讨了碳水化合物饮食(CHD)刺激下胰岛素抵抗驱动的肝脏脂质沉积的调节机制。在 ZIR 模型中,胰岛素抵抗通过提高脂肪生成酶的基因/蛋白质表达来诱导高脂血症和肝脏脂质沉积增加,脂肪生成酶是由碳水化合物反应元件结合蛋白 (ChREBP) 而不是甾醇调节元件结合蛋白 1c (SREBP-1c) 激活的。斑马鱼的代谢组学分析和小鼠肝细胞中chrebp的沉默表明,肝脏果糖 6-磷酸 (F6P) 和葡萄糖 6-磷酸 (G6P) 的增加促进了 ChREBP 介导的脂质沉积。我们进一步确定,单独的 F6P 足以通过不依赖于 SREBP-1c 的方式激活 ChREBP 介导的脂质沉积。此外,我们还阐明了受抑制的肝磷酸果糖激酶/6-磷酸葡萄糖酶功能和由葡萄糖转运蛋白 2 (GLUT2) 保留的正常葡萄糖激酶功能操纵了 ZIR 中 F6P/G6P 含量的增加。总之,本研究表明胰岛素抵抗通过 F6P/G6P 介导的 ChREBP 激活促进肝脏脂质沉积。我们的研究结果破译了胰岛素抵抗状态下肝脏脂质沉积的主要调节途径,并确定 F6P 是 ChREBP 的新潜在调节剂。
































 京公网安备 11010802027423号
京公网安备 11010802027423号