Nature Communications ( IF 14.7 ) Pub Date : 2023-09-22 , DOI: 10.1038/s41467-023-41602-1 Valdemaras Petrosius 1 , Pedro Aragon-Fernandez 1 , Nil Üresin 1, 2, 3 , Gergo Kovacs 1 , Teeradon Phlairaharn 1, 4, 5, 6 , Benjamin Furtwängler 1, 2, 3 , Jeff Op De Beeck 7 , Sarah L Skovbakke 1 , Steffen Goletz 1 , Simon Francis Thomsen 8 , Ulrich Auf dem Keller 1 , Kedar N Natarajan 1 , Bo T Porse 2, 3, 9 , Erwin M Schoof 1
|
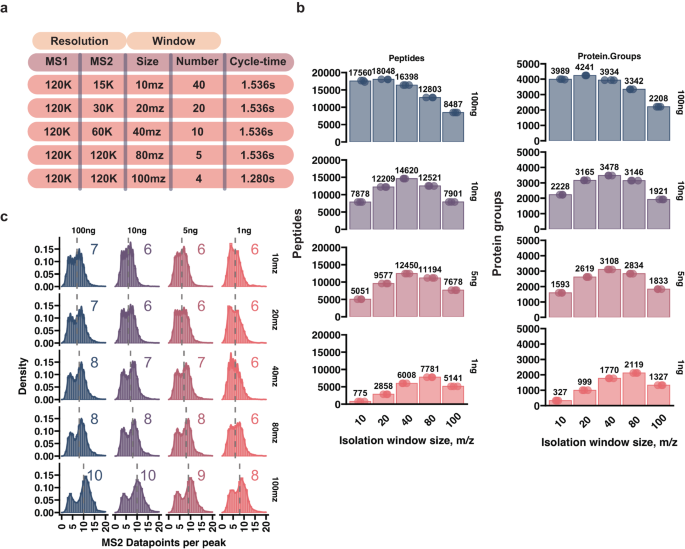
Single-cell resolution analysis of complex biological tissues is fundamental to capture cell-state heterogeneity and distinct cellular signaling patterns that remain obscured with population-based techniques. The limited amount of material encapsulated in a single cell however, raises significant technical challenges to molecular profiling. Due to extensive optimization efforts, single-cell proteomics by Mass Spectrometry (scp-MS) has emerged as a powerful tool to facilitate proteome profiling from ultra-low amounts of input, although further development is needed to realize its full potential. To this end, we carry out comprehensive analysis of orbitrap-based data-independent acquisition (DIA) for limited material proteomics. Notably, we find a fundamental difference between optimal DIA methods for high- and low-load samples. We further improve our low-input DIA method by relying on high-resolution MS1 quantification, thus enhancing sensitivity by more efficiently utilizing available mass analyzer time. With our ultra-low input tailored DIA method, we are able to accommodate long injection times and high resolution, while keeping the scan cycle time low enough to ensure robust quantification. Finally, we demonstrate the capability of our approach by profiling mouse embryonic stem cell culture conditions, showcasing heterogeneity in global proteomes and highlighting distinct differences in key metabolic enzyme expression in distinct cell subclusters.
中文翻译:
通过灵敏度定制的数据独立采集,利用单细胞蛋白质组学探索细胞状态异质性
复杂生物组织的单细胞分辨率分析对于捕获细胞状态异质性和独特的细胞信号传导模式至关重要,而基于群体的技术仍然模糊这些信号传导模式。然而,单个细胞中封装的材料数量有限,给分子分析带来了重大的技术挑战。由于广泛的优化工作,质谱单细胞蛋白质组学 (scp-MS) 已成为一种强大的工具,可通过超低的输入量进行蛋白质组分析,尽管需要进一步开发才能充分发挥其潜力。为此,我们对有限材料蛋白质组学基于轨道阱的数据独立采集(DIA)进行了全面分析。值得注意的是,我们发现高负载和低负载样本的最佳 DIA 方法之间存在根本差异。我们依靠高分辨率 MS1 定量进一步改进我们的低输入 DIA 方法,从而通过更有效地利用可用质量分析器时间来提高灵敏度。借助我们超低输入定制的 DIA 方法,我们能够适应长注射时间和高分辨率,同时保持足够低的扫描周期时间以确保稳健的定量。最后,我们通过分析小鼠胚胎干细胞培养条件、展示整体蛋白质组的异质性并强调不同细胞亚群中关键代谢酶表达的明显差异来证明我们的方法的能力。






























 京公网安备 11010802027423号
京公网安备 11010802027423号