Scientific Reports ( IF 3.8 ) Pub Date : 2023-09-08 , DOI: 10.1038/s41598-023-41109-1
Qierra R Brockman 1, 2 , Amanda Scherer 1 , Gavin R McGivney 1, 3, 4 , Wade R Gutierrez 1, 2, 3, 5 , Jeffrey Rytlewski 1 , Alexa Sheehan 1 , Akshaya Warrier 3, 5 , Emily A Laverty 1 , Grace Roughton 1 , Nina C Carnevale 1 , Vickie Knepper-Adrian 1 , Rebecca D Dodd 1, 2, 3
|
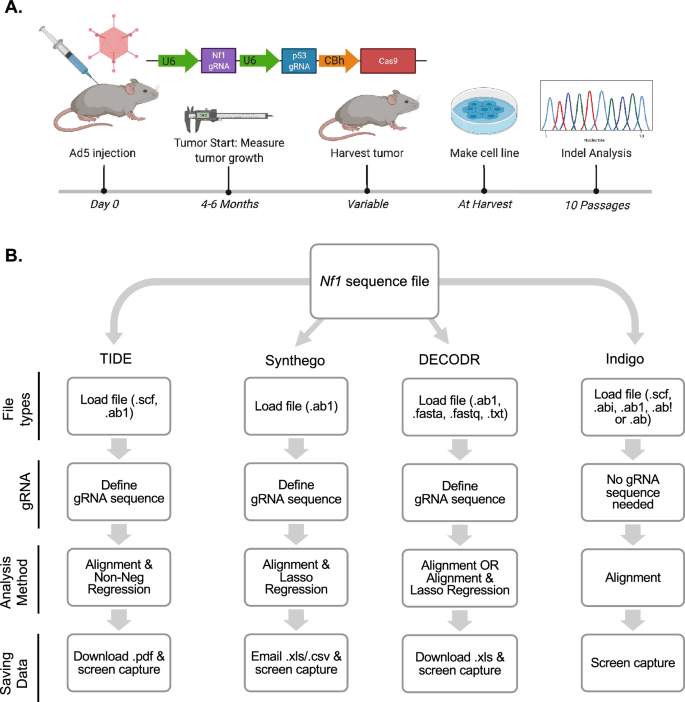
CRISPR/Cas9 gene editing has evolved from a simple laboratory tool to a powerful method of in vivo genomic engineering. As the applications of CRISPR/Cas9 technology have grown, the need to characterize the breadth and depth of indels generated by editing has expanded. Traditionally, investigators use one of several publicly-available platforms to determine CRISPR/Cas9-induced indels in an edited sample. However, to our knowledge, there has not been a cross-platform comparison of available indel analysis software in samples generated from somatic in vivo mouse models. Our group has pioneered using CRISPR/Cas9 to generate somatic primary mouse models of malignant peripheral nerve sheath tumors (MPNSTs) through genetic editing of Nf1. Here, we used sequencing data from the in vivo editing of the Nf1 gene in our CRISPR/Cas9 tumorigenesis model to directly compare results across four different software platforms. By analyzing the same genetic target across a wide panel of cell lines with the same sequence file, we are able to draw systematic conclusions about the differences in these software programs for analysis of in vivo-generated indels. Surprisingly, we report high variability in the reported number, size, and frequency of indels across each software platform. These data highlight the importance of selecting indel analysis platforms specific to the context that the gene editing approach is being applied. Taken together, this analysis shows that different software platforms can report widely divergent indel data from the same sample, particularly if larger indels are present, which are common in somatic, in vivo CRISPR/Cas9 tumor models.
中文翻译:
体细胞 CRISPR/Cas9 肿瘤发生模型的 indel 软件分辨率的差异
CRISPR/Cas9 基因编辑已经从一种简单的实验室工具发展成为一种强大的体内基因组工程方法。随着 CRISPR/Cas9 技术应用的增长,表征编辑生成的插入缺失的广度和深度的需求已经扩大。传统上,研究人员使用几个公开可用的平台之一来确定编辑样本中 CRISPR/Cas9 诱导的插入缺失。然而,据我们所知,还没有对体细胞体内小鼠模型生成的样本中可用的插入缺失分析软件进行跨平台比较。我们小组率先使用 CRISPR/Cas9 通过对 Nf1 进行基因编辑生成恶性周围神经鞘瘤 (MPNST) 的体细胞原代小鼠模型。在这里,我们使用了 CRISPR/Cas9 肿瘤发生模型中 Nf1 基因体内编辑的测序数据,直接比较了四个不同软件平台的结果。通过使用相同的序列文件分析一组细胞系中的相同遗传靶标,我们能够得出关于这些软件程序差异的系统结论,以分析体内产生的插入缺失。令人惊讶的是,我们报告的每个软件平台上报告的插入缺失数量、大小和频率都存在很大差异。这些数据强调了根据基因编辑方法的应用环境选择插入缺失分析平台的重要性。综上所述,该分析表明,不同的软件平台可以报告来自同一样本的差异很大的插入缺失数据,特别是如果存在较大的插入缺失,这在体细胞体内 CRISPR/Cas9 肿瘤模型中很常见。
































 京公网安备 11010802027423号
京公网安备 11010802027423号